Tetrahydrobiopterin deficiency
Tetrahydrobiopterin deficiency (THBD, BH4D) is a rare metabolic disorder that increases the blood levels of phenylalanine. Phenylalanine is an amino acid obtained normally through the diet, but can be harmful if excess levels build up, causing intellectual disability and other serious health problems. In healthy individuals, it is metabolised (hydroxylated) into tyrosine, another amino acid, by phenylalanine hydroxylase. However, this enzyme requires tetrahydrobiopterin as a cofactor and thus its deficiency slows phenylalanine metabolism.
| Tetrahydrobiopterin deficiency | |
|---|---|
| Other names | THB or BH4 deficiency |
-Tetrahydrobiopterin_structure.svg.png.webp) | |
| Tetrahydrobiopterin | |
| Specialty | Endocrinology |
High levels of phenylalanine are present from infancy in people with untreated tetrahydrobiopterin (THB, BH4) deficiency. The resulting signs and symptoms range from mild to severe. Mild complications may include temporary low muscle tone. Severe complications include intellectual disability, movement disorders, difficulty swallowing, seizures, behavioral problems, progressive problems with development, and an inability to control body temperature.
It was first characterized in 1975.[1]
Signs and symptoms
Genetics
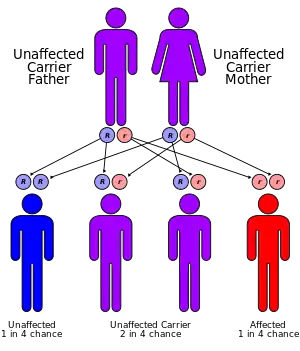
This condition is inherited in an autosomal recessive pattern, which means two copies of a specific gene in each cell are altered in order for the individual to be afflicted. Most often, the parents of an individual with an autosomal recessive disorder are carriers of one copy of the altered gene but do not show signs and symptoms of the disorder.
Related genes
Mutations in the GCH1, PCBD1, PTS and QDPR genes directly cause BH4 deficiency. Additionally, mutations of the MTHFR gene (A1298C variant) and DHFR can interfere with the recycling of BH4 and lead to less severe, but still clinifically significant, deficiencies of BH4.
BH4 is a compound that helps convert several amino acids, including phenylalanine, to other essential molecules in the body. It is also involved in the production of serotonin, dopamine, epinephrine, and norepinephrine – neurotransmitters that transmit signals between nerve cells in the brain. THB deficiency can be caused by mutations in one of several genes, including GCH1, PCBD1, PTS, and QDPR. These genes control the production of hydroxylase enzymes that are critical for producing and recycling THB. If just one of the enzymes fails to function correctly because of a gene mutation, little or no BH4 is produced. As a result, phenylalanine from the diet builds up in the bloodstream and other tissues, potentially damaging nerve cells in the brain. Tetrahydrobiopterin deficiency also disrupts the levels of certain neurotransmitters in the brain, which affects the function of the central nervous system (CNS), and dysregulation of the nitric oxide cycle leads to a buildup of peroxynitrite, an inflammatory oxidant that further degrades BH4 and perpetuates a state of inflammation.
Pathophysiology
Tetrahydrobiopterin deficiency can be caused by a deficiency of the enzyme dihydrobiopterin reductase (DHPR), whose activity is needed to replenish quinonoid-dihydrobiopterin back into its tetrahydrobiopterin form.[1] Those with this deficiency may produce sufficient levels of the enzyme phenylalanine hydroxylase (PAH) but, since tetrahydrobiopterin is a cofactor for PAH activity, deficient dihydrobiopterin reductase renders any PAH produced unable to use phenylalanine to produce tyrosine. Tetrahydrobiopterin is a cofactor in the production of L-DOPA from tyrosine and 5-hydroxy-L-tryptophan from tryptophan, which must be supplemented as treatment in addition to the supplements for classical PKU.
Other underlying causes of tetrahydrobiopterin deficiency are:[2]
Diagnosis
Directed by screening newborn for elevated plasma levels of phenyl alanine. The normal level of phenyl alanine in plasma is 1-2 mg/dl and in PKU, it normally ranges between 20-65 mg/dl. The main test for confirmation of PKU is the Guthrie test, which is a Bacillus subtilis Bioassay.
Phenyl pyruvate in urine can also be detected using Ferric Chloride test.
Treatment
Treatment of THB deficiencies consists of THB supplementation (2–20 mg/kg per day) or diet to control blood phenylalanine concentration and replacement therapy with neurotransmitters precursors (L-DOPA and 5-HTP) and supplements of folinic acid in DHPR deficiency.[3]
Tetrahydrobiopterin is available as a tablet for oral administration in the form of tetrahydrobiopterin dihydrochloride (BH4*2HCL).[4] BH4*2HCL is FDA approved under the trade name Kuvan. The typical cost of treating a patient with Kuvan is $100,000 per year.[5] BioMarin holds the patent for Kuvan until at least 2024, but Par Pharmaceutical has a right to produce a generic version by 2020.[6] BH4*2HCL is indicated at least in tetrahydrobiopterin deficiency caused by GTPCH deficiency or PTPS deficiency.[2]
Epidemiology
This condition is very rare; approximately 600 cases have been reported worldwide.[7] In most parts of the world, only 1% to 2% of all infants with high phenylalanine levels have this disorder. In Taiwan, about 30% of newborns with elevated levels of phenylalanine have a deficiency of THB.[8]
Subclinical deficiency can be found in individuals with poor diet (including low intake of folate or vitamin C) or genetic mutations in the MTHFR genes, which are involved in BH4 synthesis and recycling.
See also
- Phenylketonuria (PKU)
- Tetrahydrobiopterin (THB, BH4)
- Mild non-BH4-deficient hyperphenylalaninemia - disorder arising due to mutations of the DNAJC12 gene.
References
- Ponzone A, Spada M, Ferraris S, Dianzani I, de Sanctis L (2004). "Dihydropteridine reductase deficiency in man: from biology to treatment". Med Res Rev. 24 (2): 127–50. doi:10.1002/med.10055. PMID 14705166. S2CID 35797603.
- "Tetrahydrobiopterin Deficiency". National Organization for Rare Disorders (NORD). Retrieved 2017-10-09.
- Shintaku H (2002). "Disorders of tetrahydrobiopterin metabolism and their treatment". Curr Drug Metab. 3 (2): 123–31. doi:10.2174/1389200024605145. PMID 12003346.
- Schaub J, Däumling S, Curtius HC, Niederwieser A, Bartholomé K, Viscontini M, Schircks B, Bieri JH (1978). "Tetrahydrobiopterin therapy of atypical phenylketonuria due to defective dihydrobiopterin biosynthesis". Arch. Dis. Child. 53 (8): 674–6. doi:10.1136/adc.53.8.674. PMC 1545051. PMID 708106.
- Matthew Herper (2016-07-28). "How Focusing On Obscure Diseases Made BioMarin A $15 Billion Company". Forbes. Retrieved 2017-10-09.
- "BioMarin Announces Kuvan (sapropterin dihydrochloride) Patent Challenge Settlement". PR Newswire. 2017-04-13. Retrieved 2017-10-09.
- "BIODEF database". Retrieved September 20, 2010.
- Liu TT, Chiang SH, Wu SJ, Hsiao KJ (2001). "Tetrahydrobiopterin-deficient hyperphenylalaninemia in the Chinese". Clin Chim Acta. 313 (1–2): 157–69. doi:10.1016/S0009-8981(01)00669-6. PMID 11694255.