Ornithine transcarbamylase deficiency
Ornithine transcarbamylase deficiency also known as OTC deficiency is the most common urea cycle disorder in humans. Ornithine transcarbamylase, the defective enzyme in this disorder, is the final enzyme in the proximal portion of the urea cycle, responsible for converting carbamoyl phosphate and ornithine into citrulline. OTC deficiency is inherited in an X-linked recessive manner, meaning males are more commonly affected than females.
| Ornithine transcarbamylase deficiency | |
|---|---|
| Other names | OTC deficiency, Ornithine Carbamoyltransferase Deficiency Disease. |
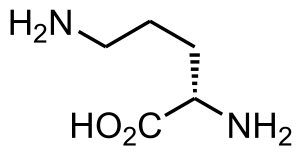 | |
| Structure of L-Ornithine | |
| Specialty | Medical genetics, metabolic syndrome, pediatrics |
| Symptoms | Tachypnea, vomiting, lethargy, loss of appetite, early morning headaches, and confusion.[1] |
| Complications | Liver failure, severe hyperammonemic encephalopathy, coma, death, intellectual, and physical disabilities.[1] |
| Causes | Genetic mutation.[1] |
| Diagnostic method | Liver function tests, genetic testing, and a liver biopsy.[1] |
| Differential diagnosis | urea cycle disorders, fulminant hepatitis, Citrin deficiency, and hyperornithinemia-hyperammonemia-homocitrullinuria syndrome.[2] |
| Treatment | Hydration, arginine, and hemodialysis.[1] |
| Prognosis | 50% of infants with OTC deficiency die.[1] |
| Frequency | 1 in 14,000 to 1 in 77,000 people.[3] |
In severely affected individuals, ammonia concentrations increase rapidly causing ataxia, lethargy and death without rapid intervention. OTC deficiency is diagnosed using a combination of clinical findings and biochemical testing, while confirmation is often done using molecular genetics techniques.
Once an individual has been diagnosed, the treatment goal is to avoid precipitating episodes that can cause an increased ammonia concentration. The most common treatment combines a low protein diet with nitrogen scavenging agents. Liver transplant is considered curative for this disease. Experimental trials of gene therapy using adenoviral vectors resulted in the death of one participant, Jesse Gelsinger, and have been discontinued.
Signs and symptoms
As with several other metabolic conditions, OTC deficiency can have variable presentations, regarding age of onset and the severity of symptoms. This compounded when considering heterozygous females and the possibility of non-random X-inactivation. In the classic and most well-known presentation, a male infant appears well initially, but by the second day of life they are irritable, lethargic and stop feeding. A metabolic encephalopathy develops, and this can progress to coma and death without treatment.[4] Ammonia is only toxic to the brain, other tissues can handle elevated ammonia concentrations without problems.[5]
Later onset forms of OTC deficiency can have variable presentations. Although late onset forms of the disease are often considered milder than the classic infantile presentation, any affected individual is at risk for an episode of hyperammonemia that could still be life-threatening, if presented with the appropriate stressors.[6] These patients will often present with headaches, nausea, vomiting, delayed growth and a variety of psychiatric symptoms (confusion, delirium, aggression, or self-injury).[5] A detailed dietary history of an affected individual with undiagnosed OTC deficiency will often reveal a history of protein avoidance.[6]
The prognosis of a patient with severe OTC deficiency is well correlated with the length of the hyperammonemic period rather than the degree of hyperammonemia or the presence of other symptoms, such as seizures.[6] Even for patients with late onset forms of the disease, their overall clinical picture is dependent on the extent of hyperammonemia they have experienced, even if it has remained unrecognized.[5]
Genetics
OTC deficiency is caused by mutations in the OTC gene, which is located on the X chromosome.[7] OTC codes for the mitochondrial enzyme ornithine transcarbamylase, which is expressed only in liver. The functional enzyme consists of three identical subunits.[8] OTC is the last enzyme in the proximal portion of the urea cycle, which consists of the reactions that take place in the mitochondria. The substrates of the reaction catalyzed by ornithine transcarbamylase are ornithine and carbamyl phosphate, while the product is citrulline.[5]
There are no common mutations that cause disease, however 10 - 15% of disease causing mutations are deletions.[7] It is inherited in an X-linked recessive manner, meaning males are more commonly affected than females. Females who carry a defective copy of the gene can be severely affected or asymptomatic, largely depending on the random nature of X-inactivation.[7] There is some degree of genotype — phenotype correlation with OTC deficiency, but this depends on a number of situations. Individuals with milder mutations, often associated with late onset disease can still present with severe illness when exposed to sufficient metabolic stress. Correlations are more difficult to ascertain in females, since the residual activity of OTC in the liver is impacted not only by the nature of the mutation, but also by the random pattern of X-inactivation.[6] OTC deficiency is estimated to be the most common urea cycle disorder.[6] An exact incidence is difficult to calculate, due to the varying clinical presentations of later onset forms of the disease. Early estimates of the incidence were as high as 1:14,000 live births, however later studies have decreased these estimates to approximately 1:60,000 - 1:72,000.[6]
Diagnosis
In individuals with marked hyperammonemia, a urea cycle disorder is usually high on the list of possible causes. While the immediate focus is lowering the patient's ammonia concentrations, identifying the specific cause of increased ammonia levels is key as well.
Diagnostic testing for OTC deficiency, or any individual with hyperammonemia involves plasma and urine amino acid analysis, urine organic acid analysis (to identify the presence or absence of orotic acid, as well as rule out an organic acidemia) and plasma acylcarnitines (will be normal in OTC deficiency, but can identify some other causes of hyperammonemia). An individual with untreated OTC deficiency will show decreased citrulline and arginine concentrations (because the enzyme block is proximal to these intermediates) and increased orotic acid.[4] The increased orotic acid concentrations result from the buildup of carbamoyl phosphate. This biochemical phenotype (increased ammonia, low citrulline and increased orotic acid) is classic for OTC deficiency, but can also be seen in neonatal presentations of ornithine aminotransferase deficiency.[4] Only severely affected males consistently demonstrate this classic biochemical phenotype.
Heterozygous females can be difficult to diagnose. With the rise of sequencing techniques, molecular testing has become preferred, particularly when the disease causing mutations in the family are known.[7] Historically, heterozygous females were often diagnosed using an allopurinol challenge. In a female with reduced enzyme activity, an oral dose of allopurinol would be metabolized to oxypurinol ribonucleotide, which blocks the pyrimidine biosynthetic pathway. When this induced enzymatic block is combined with reduced physiologic enzyme activity as seen in heterozygotes, the elevation of orotic acid could be used to differentiate heterozygotes from unaffected individuals. This test was not universally effective, as it had both false negative and false positive results.[5]
Ornithine transcarbamylase is only expressed in the liver, thus performing an enzyme assay to confirm the diagnosis requires a liver biopsy. Before molecular genetic testing was commonly available, this was one of the only methods for confirmation of a suspected diagnosis. In cases where prenatal diagnosis was requested, a fetal liver biopsy used to be required to confirm if a fetus was affected.[4] Modern molecular techniques have eliminated this need, and gene sequencing is now the preferred method of diagnosis in asymptomatic family members after the diagnosis has been confirmed in a proband.[6][7]
Treatment
The treatment goal for individuals affected with OTC deficiency is the avoidance of hyperammonemia. This can be accomplished through a strictly controlled low-protein diet, as well as preventative treatment with nitrogen scavenging agents such as sodium benzoate. The goal is to minimize the nitrogen intake while allowing waste nitrogen to be excreted by alternate pathways.[7] Arginine is typically supplemented as well, in an effort to improve the overall function of the urea cycle.[7] If a hyperammonemic episode occurs, the aim of treatment is to reduce the individual's ammonia levels as soon as possible. In extreme cases, this can involve hemodialysis.[4]
Gene therapy had been considered a possibility for curative treatment for OTC deficiency, and clinical trials were taking place at the University of Pennsylvania in the late 1990s. These were halted after the death of Jesse Gelsinger, a young man taking part in a phase I trial using an adenovirus vector.[9] Currently, the only option for curing OTC deficiency is a liver transplant, which restores normal enzyme activity.[10] A 2005 review of 51 patients with OTC deficiency who underwent liver transplant estimated 5-year survival rates of greater than 90%.[10] Severe cases of OTC deficiency are typically evaluated for liver transplant by 6 months of age.[6]
Prognosis
A 1999 retrospective study of 74 cases of neonatal onset found that 32 (43%) patients died during their first hyperammonemic episode. Of those who survived, less than 20% survived to age 14. Few of these patients received liver transplants.[11]
In pop culture
Ornithine transcarbamylase deficiency was the final diagnosis of a patient treated in the 15th episode of 1st season of House, M. D. It was also the final diagnosis of a patient that died in the 3rd episode of 3rd season of Chicago Med.
References
- Donovan, Kathleen; Guzman, Nilmarie (August 8, 2022). "Ornithine Transcarbamylase Deficiency". StatPearls Publishing. PMID 30725942. Retrieved October 22, 2023.
- Lichter-Konecki, Uta; Caldovic, Ljubica; Morizono, Hiroki; Simpson, Kara; Mew, Nicholas Ah; MacLeod, Erin (May 26, 2022). "Ornithine Transcarbamylase Deficiency". University of Washington, Seattle. PMID 24006547. Retrieved October 22, 2023.
- "Ornithine transcarbamylase deficiency: MedlinePlus Genetics". MedlinePlus. October 1, 2017. Retrieved October 22, 2023.
- Wraith, J. E. (2001). "Ornithine carbamoyltransferase deficiency". Archives of Disease in Childhood. 84 (1): 84–88. doi:10.1136/adc.84.1.84. PMC 1718609. PMID 11124797.
- Walker, V. (2009). "Ammonia toxicity and its prevention in inherited defects of the urea cycle". Diabetes, Obesity and Metabolism. 11 (9): 823–835. doi:10.1111/j.1463-1326.2009.01054.x. PMID 19531057. S2CID 25998574.
- Lichter-Konecki, U.; Caldovic, L.; Morizono, H.; Simpson, K.; Pagon, R. A.; Adam, M. P.; Bird, T. D.; Dolan, C. R.; Fong, C. T.; Stephens, K. (1993). "Ornithine Transcarbamylase Deficiency". PMID 24006547.
{{cite journal}}: Cite journal requires|journal=(help) - "#311250 - Ornithine Transcarbamylase Deficiency, Hyperammonemia Due To". Johns Hopkins University. Retrieved 2014-01-01.
- "Human ornithine transcarbamylase (OTC) mRNA, complete coding sequence". US National Library of Medicine.
{{cite web}}: Missing or empty|url=(help) - Deakin, C. T.; Alexander, I. E.; Kerridge, I. (2009). "Accepting Risk in Clinical Research: Is the Gene Therapy Field Becoming Too Risk-averse?". Molecular Therapy. 17 (11): 1842–1848. doi:10.1038/mt.2009.223. PMC 2835028. PMID 19773741.
- Morioka, D.; Kasahara, M.; Takada, Y.; Shirouzu, Y.; Taira, K.; Sakamoto, S.; Uryuhara, K.; Egawa, H.; Shimada, H.; Tanaka, K. (2005). "Current role of liver transplantation for the treatment of urea cycle disorders: A review of the worldwide English literature and 13 cases at Kyoto University". Liver Transplantation. 11 (11): 1332–1342. doi:10.1002/lt.20587. PMID 16237708. S2CID 25787334.
- Maestri, N. E.; Clissold, D.; Brusilow, S. W. (1999-03-01). "Neonatal onset ornithine transcarbamylase deficiency: A retrospective analysis". The Journal of Pediatrics. 134 (3): 268–272. doi:10.1016/s0022-3476(99)70448-8. ISSN 0022-3476. PMID 10064660.