Ethylmalonic encephalopathy
Ethylmalonic encephalopathy (EE) is a rare autosomal recessive inborn error of metabolism. Patients affected with EE are typically identified shortly after birth, with symptoms including diarrhea, petechiae and seizures.[1][2] The genetic defect in EE is thought to involve an impairment in the degradation of sulfide intermediates in the body. Hydrogen sulfide then builds up to toxic levels.[3] EE was initially described in 1994.[4] Most cases of EE have been described in individuals of Mediterranean or Arabic origin.[3]
| Ethylmalonic encephalopathy | |
|---|---|
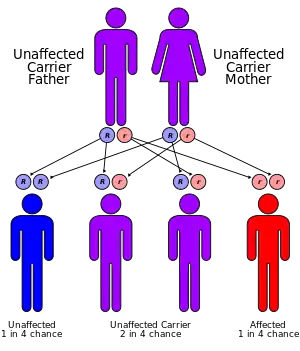 | |
| Ethylmalonic encephalopathy has an autosomal recessive pattern of inheritance. | |
| Specialty | Medical genetics |
Signs and symptoms
Neurologic signs and symptoms include progressively delayed development, weak muscle tone (hypotonia), seizures, and abnormal movements. The body's network of blood vessels is also affected. Children with this disorder may experience rashes of tiny red spots (petechiae) caused by bleeding under the skin and blue discoloration in the hands and feet due to reduced oxygen in the blood (acrocyanosis). Chronic diarrhea is another common feature of ethylmalonic encephalopathy.[3] EE is often identified by urine organic acid analysis, the excretion of ethylmalonic acid, methylsuccinic acid, isobutyrylglycine and isovalerylglucine. Patients will also often have elevated thiosulphate concentration in their urine.[5]
The signs and symptoms of ethylmalonic encephalopathy are apparent at birth or begin in the first few months of life. Problems with the nervous system typically worsen over time, and most affected individuals survive only into early childhood. A few children with a milder, chronic form of this disorder have been reported, and there can be considerable phenotypic variation, even within families.[6] The life expectancy of individuals with EE is less than ten years.[3]
Pathophysiology
Mutations in the ETHE1 gene cause ethylmalonic encephalopathy.[7] The ETHE1 gene makes an enzyme that plays an important role in energy production. It is active in mitochondria, which are the energy-producing centers within cells. Little is known about its exact function, however.
Mutations in the ETHE1 gene lead to the production of a defective version of the enzyme or prevents the enzyme from being made. A lack of the ETHE1 enzyme impairs the ability to make energy in mitochondria. Additionally, a loss of this enzyme allows potentially toxic compounds, including ethylmalonic acid and lactic acid, to build up in the body. Excess amounts of these compounds can be detected in urine. It remains unclear how a loss of the ETHE1 enzyme leads to progressive brain dysfunction and the other features of ethylmalonic encephalopathy.
Ethylmalonic encephalopathy is an autosomal recessive disorder, which means the defective gene is located on an autosome, and both parents must carry one copy of the defective gene in order to have a child born with the disorder. The parents of a child with an autosomal recessive disorder are usually not affected by the disorder.
Diagnosis
References
- Zafeiriou DI, Augoustide-Savvopoulou P, Haas D, Smet J, Triantafyllou P, Vargiami E, Tamiolaki M, Gombakis N, van Coster R, Seweil AC, Vianey-Saban C, Gregersen N (2007). "Ethylmalonic encephalopathy: clinical and biochemical observations". Neuropediatrics. 38 (2): 78–82. doi:10.1055/s-2007-984447. PMID 17712735.
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; Zeviani, M. (2009). "Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy". Nature Medicine. 15 (2): 200–205. doi:10.1038/nm.1907. PMID 19136963. S2CID 5970257.
- "Encephalopathy, Ethylmalonic". Johns Hopkins University. Retrieved 2012-05-12.
- Burlina, A. B.; Dionisi-Vici, C.; Bennett, M. J.; Gibson, K. M.; Servidei, S.; Bertini, E.; Hale, D. E.; Schmidt-Sommerfeld, E.; Sabetta, G.; Zacchello, F.; Rinaldo, P. (1994). "A new syndrome with ethylmalonic aciduria and normal fatty acid oxidation in fibroblasts". The Journal of Pediatrics. 124 (1): 79–86. doi:10.1016/S0022-3476(94)70257-8. PMID 8283379.
- Drousiotou, A.; Dimeo, I.; Mineri, R.; Georgiou, T.; Stylianidou, G.; Tiranti, V. (2011). "Ethylmalonic encephalopathy: Application of improved biochemical and molecular diagnostic approaches". Clinical Genetics. 79 (4): 385–390. doi:10.1111/j.1399-0004.2010.01457.x. PMID 20528888. S2CID 42297374.
- Pigeon, N.; Campeau, P. M.; Cyr, D.; Lemieux, B.; Clarke, J. T. R. (2009). "Clinical Heterogeneity in Ethylmalonic Encephalopathy". Journal of Child Neurology. 24 (8): 991–996. doi:10.1177/0883073808331359. PMID 19289697. S2CID 10613701.
- Mineri R, Rimoldi M, Burlina AB, Koskull S, Perletti C, Heese B, Von Döbeln U, Mereghetti P, Di Meo I, Invernizzi F, Zeviani M, Uziel G, Tiranti V (Jul 2008). "Identification of new mutations in the ETHE1 gene in a cohort of 14 patients presenting with ethylmalonic encephalopathy". Journal of Medical Genetics. 45 (7): 473–8. doi:10.1136/jmg.2008.058271. PMID 18593870. S2CID 42504096.
External links
- Ethylmalonic encephalopathy at NLM Genetics Home Reference