Nucleotide excision repair
Nucleotide excision repair is a DNA repair mechanism.[2] DNA damage occurs constantly because of chemicals (e.g. intercalating agents), radiation and other mutagens. Three excision repair pathways exist to repair single stranded DNA damage: Nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR). While the BER pathway can recognize specific non-bulky lesions in DNA, it can correct only damaged bases that are removed by specific glycosylases. Similarly, the MMR pathway only targets mismatched Watson-Crick base pairs.
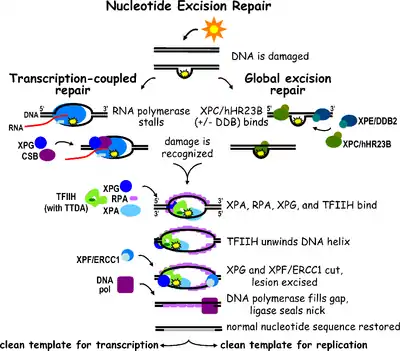
Nucleotide excision repair (NER) is a particularly important excision mechanism that removes DNA damage induced by ultraviolet light (UV). UV DNA damage results in bulky DNA adducts - these adducts are mostly thymine dimers and 6,4-photoproducts. Recognition of the damage leads to removal of a short single-stranded DNA segment that contains the lesion. The undamaged single-stranded DNA remains and DNA polymerase uses it as a template to synthesize a short complementary sequence. Final ligation to complete NER and form a double stranded DNA is carried out by DNA ligase. NER can be divided into two subpathways: global genomic NER (GG-NER or GGR) and transcription coupled NER (TC-NER or TCR). The two subpathways differ in how they recognize DNA damage but they share the same process for lesion incision, repair, and ligation.
The importance of NER is evidenced by the severe human diseases that result from in-born genetic mutations of NER proteins. Xeroderma pigmentosum and Cockayne's syndrome are two examples of NER associated diseases.
In eukaryotes
Nucleotide excision repair is more complex in eukaryotes than prokaryotes, but the general principle is similar. There are 9 major proteins involved in NER in mammalian cells. Deficiencies in certain proteins leads to disease; protein names are associated with the disease. XPA, XPB, XPC, XPD, XPE, XPF, and XPG all derive from хeroderma pigmentosum and CSA and CSB represent proteins linked to Cockayne syndrome. Additionally, the proteins ERCC1, RPA, RAD23A, RAD23B, and others also participate in nucleotide excision repair. A more complete list of proteins involved in NER is found below.
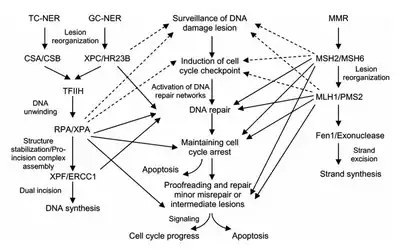
Eukaryotic nucleotide excision repair can be divided into two subpathways: global genomic NER (GG-NER) and transcription coupled NER (TC-NER). Three different sets of proteins are involved in recognizing DNA damage for each subpathway. After damage recognition, the three subpathways converge for the steps of dual incision, repair, and ligation.
Global genomic NER (GG-NER)
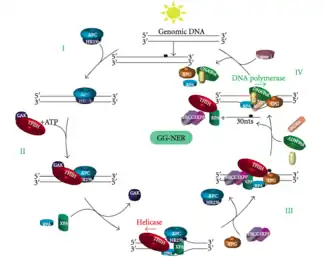
Global genomic NER repairs damage in both transcribed and untranscribed DNA strands in active and inactive genes throughout the genome. This process is not dependent on transcription. This pathway employs several "damage sensing" proteins including the DNA-damage binding (DDB) and XPC-Rad23B complexes that constantly scan the genome and recognize helix distortions: the XPC-Rad23B complex is responsible for distortion recognition, while DDB1 and DDB2 (XPE) can also recognize some types of damage caused by UV light. Additionally, XPA performs a function in damage recognition that is as yet poorly defined. Upon identification of a damaged site, subsequent repair proteins are then recruited to the damaged DNA to verify presence of DNA damage, excise the damaged DNA surrounding the lesion then fill in the repair patch.
GG-NER associated diseases
Mutations in GG-NER machinery are responsible for multiple genetic disorders including:
- Xeroderma pigmentosum (XP): severe photosensitivity, high cancer rates in areas of the body exposed to the sun (e.g. skin)
Transcription coupled repair (TC-NER)
At any given time, most of the genome in an organism is not undergoing transcription; there is a difference in NER efficiency between transcriptionally silent and transcriptionally active regions of the genome. For many types of lesions, NER repairs the transcribed strands of transcriptionally active genes faster than it repairs nontranscribed strands and transcriptionally silent DNA.
TC-NER and GG-NER differ only in the initial steps of DNA damage recognition. The principal difference between TC-NER and GG-NER is that TC-NER does not require XPC or DDB proteins for distortion recognition in mammalian cells. Instead TC-NER initiates when RNA polymerase stalls at a lesion in DNA: the blocked RNA polymerase serves as a damage recognition signal, which replaces the need for the distortion recognition properties of the XPC-RAD23B and DDB complexes. CS proteins (CSA and CSB) bind some types of DNA damage instead of XPC-Rad23B.
Other repair mechanisms are possible but less accurate and efficient.
TC-NER associated diseases
TC-NER initiates when RNA polymerase stalls at a lesion in DNA, whereupon protein complexes help move the polymerase backwards. Mutations in TC-NER machinery are responsible for multiple genetic disorders including:
- Trichothiodystrophy (TTD): some individuals are photosensitive, ichthyosis, mental/physical retardation
- Cockayne syndrome (CS): photosensitivity, mental retardation, progeria-like features, microcephaly
Dual incision
Transcription factor II H (TFIIH) is the key enzyme involved in dual excision. TFIIH and XPG are first recruited to the site of DNA damage (XPG stabilizes TFIIH). The TFIIH subunits of XPD and XPB act as a 5'-3' and 3'-5' helicase, respectively - they help unwind DNA and generate a junction between the double-stranded and single-stranded DNA around the transcription bubble. In addition to stabilizing TFIIH, XPG also has endonuclease activity; it cuts DNA damage on the 3' side while the XPF–ERCC1 heterodimeric protein cuts on the 5' side. The dual incision leads to the removal of a ssDNA with a single strand gap of 25~30 nucleotides. The small, excised, damage-containing DNA (sedDNA) oligonucleotides are initially released from the duplex in complex with TFIIH but then dissociate in an ATP-dependent manner and become bound to replication protein A (RPA). Inhibition of gap filling DNA synthesis and ligation results in an accumulation of RPA-bound sedDNAs in the cell.
Replication protein A (RPA) and XPA are the last two proteins associated with the main NER repair complex. These two proteins are present prior to TFIIH binding since they are involved with verifying DNA damage. They may also protect single-stranded DNA. After verification, the 5' side incision is made and DNA repair begins before the 3' side incision. This helps reduce exposed single stranded DNA during the repair process.
Repair and ligation
Replication factor C (RFC) loads the Proliferating Cell Nuclear Antigen (PCNA) onto the DNA strand. This allows DNA polymerases implicated in repair (δ, ε and/or κ) to copy the undamaged strand via translocation. DNA ligase I and Flap endonuclease 1 or the Ligase-III-XRCC1 complex seal the nicks to complete NER.
In prokaryotes: Uvr proteins

The process of nucleotide excision repair is controlled in Escherichia coli by the UvrABC endonuclease enzyme complex, which consists of four Uvr proteins: UvrA, UvrB, UvrC, and DNA helicase II (sometimes also known as UvrD in this complex). First, a UvrA-UvrB complex scans the DNA, with the UvrA subunit recognizing distortions in the helix, caused for example by pyrimidine dimers. When the complex recognizes such a distortion, the UvrA subunit leaves and an UvrC protein comes in and binds to the UvrB monomer and, hence, forms a new UvrBC dimer. UvrB cleaves a phosphodiester bond 4 nucleotides downstream of the DNA damage, and the UvrC cleaves a phosphodiester bond 8 nucleotides upstream of the DNA damage and created 12 nucleotide excised segment. DNA helicase II (sometimes called UvrD) then comes in and removes the excised segment by actively breaking the hydrogen bonds between the complementary bases. The resultant gap is then filled in using DNA polymerase I and DNA ligase. The basic excision process is very similar in higher cells, but these cells usually involve many more proteins – E.coli is a simple example.[5]
TC-NER also exists in bacteria, and is mediated by the TRCF (Mfd) protein. TRCF is an SF2 ATPase that uses ATP hydrolysis to translocate on dsDNA upstream of the transcription bubble and forward translocate RNA polymerase, thus initiating dissociation of the RNA Polymerase ternary elongation complex. TRCF also recruits the Uvr(A)BC nucleotide excision repair machinery by direct physical interaction with the UvrA subunit.
Cancer
Though historical studies have shown inconsistent results, genetic variation or mutation to nucleotide excision repair genes can impact cancer risk by affecting repair efficacy. Single-nucleotide polymorphisms (SNPs) and nonsynonymous coding SNPs (nsSNPs) are present at very low levels (>1%) in the human population.[7] If located in NER genes or regulatory sequences, such mutations can negatively affect DNA repair capacity resulting in an increase likelihood of cancer development. While the functional impact of all polymorphisms has not been characterized, some polymorphisms in DNA repair genes or their regulatory sequences do induce phenotypical changes and are involved in cancer development.[8] A study of lung cancer cases found modest association between NER specific SNP polymorphisms and lung cancer risk.[9] The results indicate that some inherited polymorphic variations in NER genes may result in predisposition to lung cancer, and potentially other cancer states.
NER dysfunction result of DNA polymorphism
Two important genes in the NER pathway for which polymorphism has shown functional and phenotypic impact are the XPD and XPC genes.[10] XPD, also known as ERCC2, serves to open DNA around the site of damage during NER, in addition to other transcriptional activities. Studies have shown that polymorphisms at Exon 10 (G>A)(Asp312Asn) and Exon 23 (A>T)(Lys751Gln) are linked with genetic predisposition to several cancer types.[11][12] The XPC gene is responsible for a protein which recognizes DNA during the early portion of the NER pathway. This gene can have polymorphisms at Intron 9 and SNPs in Exon 15 which have been correlated with cancer risk as well. Research has shown that a biallelic poly (AT) insertion/deletion polymorphism in Intron 9 of XPC is associated with increased risk for skin, breast and prostate cancers,[12][13][14] especially in North Indian populations.
Impact on cancer prognosis
The study of a hereditary cancer, xeroderma pigmentosum has helped identify several genes which encode proteins in the NER pathway, two of which are XPC and XPD. XP is caused by a homozygous deficiency in UV DNA damage repair (GG-NER) which increases the patients' risk of skin cancer by 1000-fold. In heterozygous patients, the risk of cancer is sporadic but can be predicted based on analytical assessment of polymorphisms in XP related DNA repair genes purified from lymphocytes.[15] In a study relapse rates of high-risk stage II and III colorectal cancers, XPD (ERCC2) polymorphism 2251A>C was significantly correlated with early relapse after chemotherapeutic treatment.[16] Studies have indicated that the effects of polymorphic NER genes is additive, with greater frequency of variants, greater cancer risk presents.[15][16][17]
Aging
In humans and mice, germline mutation in genes employed in NER cause features of premature aging. These genes and their corresponding proteins include ERCC1(ERCC1), ERCC2(XPD), ERCC3(XPB), ERCC4(XPF), ERCC5 (XPG), ERCC6(CSB) and ERCC8(CSA).
DNA repair-deficient ERCC1 mutant mice show features of accelerated aging, and have a limited lifespan.[18] Accelerated aging in the mutant involves numerous organs.
Mutations in the ERCC2(XPD) gene can lead to various syndromes, either xeroderma pigmentosum (XP), trichothiodystrophy (TTD) or a combination of XP and TTD (XPTTD), or a combination of XP and Cockayne syndrome (XPCS).[19] TTD and CS both display features of premature aging. These features may include sensorineural deafness, retinal degeneration, white matter hypomethylation, central nervous system calcification, reduced stature, and cachexia (loss of subcutaneous fat tissue).[19][20] XPCS and TTD fibroblasts from ERCC2(XPD) mutant human and mouse show evidence of defective repair of oxidative DNA damages that may underlie the segmental progeroid (premature aging) symptoms[21] (see DNA damage theory of aging).
Mutations in the ERCC3(XPB) gene can lead, in humans, to xeroderma pigmentosum (XP) or XP combined with Cockayne syndrome (XPCS).[22]
Deficiency of ERCC4(XPF) in humans results in a variety of conditions including accelerated aging.[23]
In humans, mutational defects in the ERCC5(XPG) gene can cause either the cancer-prone condition xeroderma pigmentosum (XP) alone, or in combination with the severe neurodevelopmental disorder Cockayne syndrome (CS) or the infantile lethal cerebro-oculo-facio-skeletal syndrome.[24] An ERCC5(XPG) mutant mouse model presents features of premature aging including cachexia and osteoporosis with pronounced degenerative phenotypes in both liver and brain.[24] These mutant mice develop a multi-system premature aging degenerative phenotype that appears to strengthen the link between DNA damage and aging.[24](see DNA damage theory of aging).
Cockayne syndrome (CS) arises from germline mutations in either of two genes ERCC8(CSA) or ERCC6(CSB). ERCC8(CSA) mutations generally give rise to a more moderate form of CS than ERCC6(CSB) mutations.[25] Mutations in the CSA gene account for about 20% of CS cases.[26] Individuals with CSA and CSB are characterised by severe postnatal growth and mental retardation and accelerated aging leading to premature death at the age of 12 to 16 years.[27]
Decline in NER with aging
As reviewed by Gorbunova et al.,[28] studies of NER in different cells and tissues from young and old individuals frequently have shown a decrease in NER capacity with increasing age. This decline may be due to reduced constitutive levels of proteins employed in the NER pathway.[29]
NER associated genes
| Human Gene (Protein) | Mouse Ortholog | Yeast Ortholog | Subpathway | Function in NER | GeneCards Entry |
|---|---|---|---|---|---|
| CCNH (Cyclin H) | Ccnh | CCL1 | Both | CDK Activator Kinase (CAK) subunit | CCNH |
| CDK7 (Cyclin Dependent Kinase (CDK) 7)) | Cdk7 | KIN28 | Both | CAK subunit | CDK7 |
| CETN2 (Centrin-2) | Cetn2 | Unknown | GGR | Damage recognition; forms complex with XPC | CETN2 |
| DDB1 (DDB1) | Ddb1 | Unknown | GGR | Damage recognition; forms complex with DDB2 | DDB1 |
| DDB2 (DDB2) | Ddb2/Xpe | Unknown | GGR | Damage recognition; recruits XPC | DDB2 |
| ERCC1 (ERCC1) | Ercc1 | RAD10 | Both | Involved in incision on 3' side of damage; forms complex with XPF | ERCC1 |
| ERCC2 (XPD) | Ercc2 | RAD3 | Both | ATPase and helicase activity; transcription factor II H (TFIIH) subunit | ERCC2 |
| ERCC3 (XPB) | Ercc3 | RAD25 | Both | ATPase and helicase activity; transcription factor II H (TFIIH) subunit | ERCC3 |
| ERCC4 (XPF) | Ercc4 | RAD1 | Both | Involved in incision on 3' side of damage; structure specific endonuclease | ERCC4 |
| ERCC5 (XPG) | Ercc5 | RAD2 | Both | Involved in incision on 5' side of damage; stabilizes TFIIH; structure specific endonuclease | ERCC5 |
| ERCC6 (CSB) | Ercc6 | RAD26 | TC-NER | Transcription elongation factor; involved in transcription coupling and chromatin remodelling | ERCC6 |
| ERCC8 (CSA) | Ercc8 | RAD28 | TC-NER | Ubiquitin ligase complex; interacts with CSB and p44 of TFIIH | ERCC8 |
| LIG1 (DNA Ligase I) | Lig1 | CDC9 | Both | Final ligation | LIG1 |
| MNAT1 (MNAT1) | Mnat1 | TFB3 | Both | Stabilizes CAK complex | MNAT1 |
| MMS19 (MMS19) | Mms19 | MET18 | Both | Interacts with XPD and XPB subunits of TFIIH helicases | MMS19 |
| RAD23A (RAD23A) | Rad23a | RAD23 | GGR | Damage recognition; forms complex with XPC | RAD23A |
| RAD23B (RAD23B) | Rad23b | RAD23 | GGR | Damage recognition, forms complex with XPC | RAD23B |
| RPA1 (RPA1) | Rpa1 | RFA1 | Both | Subunit of RFA complex | RPA1 |
| RPA2 (RPA2) | Rpa2 | RFA2 | Both | Subunit of RFA complex | RPA2 |
| TFIIH (Transcription factor II H) | Gtf2h1-3 | Tfb1 Ssl1 Tfb4 | Both | Involved in incision, forms complex around lesion | GTF2H1 GTF2H2 GTF2H3 |
| XAB2 (XAB2) | Xab2 | SYF1 | TC-NER | Damage recognition; interacts with XPA, CSA, and CSB | XAB2 |
| XPA (XPA) | Xpa | RAD14 | Both | Damage recognition | XPA |
| XPC (XPC) | Xpc | RAD4 | GGR | Damage recognition | XPC |
See also
- Base excision repair (BER)
- Mismatch repair (MMR)
References
- Fuss JO, Cooper PK (June 2006). "DNA repair: dynamic defenders against cancer and aging". PLOS Biology. 4 (6): e203. doi:10.1371/journal.pbio.0040203. PMC 1475692. PMID 16752948.

- Carroll SB; Wessler SR; Griffiths AJFl; Lewontin RC (2008). Introduction to genetic analysis. New York: W.H. Freeman and Co. p. 534. ISBN 978-0-7167-6887-6.
- Le May N, Egly JM, Coin F (2010). "True lies: the double life of the nucleotide excision repair factors in transcription and DNA repair". Journal of Nucleic Acids. 2010: 1–10. doi:10.4061/2010/616342. PMC 2915888. PMID 20725631.
- Morita R, Nakane S, Shimada A, et al. (2010). "Molecular mechanisms of the whole DNA repair system: a comparison of bacterial and eukaryotic systems". Journal of Nucleic Acids. 2010: 1–32. doi:10.4061/2010/179594. PMC 2957137. PMID 20981145.
- Truglio JJ, Croteau DL, Van Houten B, Kisker C (February 2006). "Prokaryotic nucleotide excision repair: the UvrABC system". Chemical Reviews. 106 (2): 233–252. doi:10.1021/cr040471u. PMID 16464004.
- Zhang Y, Rohde LH, Wu H (June 2009). "Involvement of nucleotide excision and mismatch repair mechanisms in double strand break repair". Current Genomics. 10 (4): 250–258. doi:10.2174/138920209788488544. PMC 2709936. PMID 19949546.
- Kwok PY, Gu Z (December 1999). "Single nucleotide polymorphism libraries: why and how are we building them?". Molecular Medicine Today. 5 (12): 538–543. doi:10.1016/S1357-4310(99)01601-9. PMID 10562720.
- Karahalil B, Bohr V, Wilson D (October 2012). "Impact of DNA polymorphisms in key DNA base excision repair proteins on cancer risk". Human & Experimental Toxicology. 31 (10): 981–1005. doi:10.1177/0960327112444476. PMC 4586256. PMID 23023028.
- Sakoda LC, Loomis MM, Doherty JA, Julianto L, Barnett MJ, Neuhouser ML, Thornquist MD, Weiss NS, Goodman GE, Chen C (2012). "Germ line variation in nucleotide excision repair genes and lung cancer risk in smokers". International Journal of Molecular Epidemiology and Genetics. 3 (1): 1–17. PMC 3316453. PMID 22493747.
- Hou SM, Fält S, Angelini S, Yang K, Nyberg F, Lambert B, Hemminki K (April 2002). "The XPD variant alleles are associated with increased aromatic DNA adduct level and lung cancer risk". Carcinogenesis. 23 (4): 599–603. doi:10.1093/carcin/23.4.599. PMID 11960912.
- Wang M, Gu D, Zhang Z, Zhou J, Zhang Z (2009). "XPD polymorphisms, cigarette smoking, and bladder cancer risk: a meta-analysis". Journal of Toxicology and Environmental Health Part A. 72 (11–12): 698–705. doi:10.1080/15287390902841029. PMID 19492231. S2CID 22991719.
- Mittal RD, Mandal RK (January 2012). "Genetic variation in nucleotide excision repair pathway genes influence prostate and bladder cancer susceptibility in North Indian population". Indian Journal of Human Genetics. 18 (1): 47–55. doi:10.4103/0971-6866.96648. PMC 3385179. PMID 22754221.
- Blankenburg S, König IR, Moessner R, Laspe P, Thoms KM, Krueger U, Khan SG, Westphal G, Berking C, Volkenandt M, Reich K, Neumann C, Ziegler A, Kraemer KH, Emmert S (June 2005). "Assessment of 3 xeroderma pigmentosum group C gene polymorphisms and risk of cutaneous melanoma: a case-control study". Carcinogenesis. 26 (6): 1085–1090. doi:10.1093/carcin/bgi055. PMID 15731165.
- Shore RE, Zeleniuch-Jacquotte A, Currie D, Mohrenweiser H, Afanasyeva Y, Koenig KL, Arslan AA, Toniolo P, Wirgin I (May 2008). "Polymorphisms in XPC and ERCC2 genes, smoking and breast cancer risk". International Journal of Cancer. 122 (9): 2101–2105. doi:10.1002/ijc.23361. PMID 18196582. S2CID 9456435.
- Qiao Y, Spitz MR, Guo Z, Hadeyati M, Grossman L, Kraemer KH, Wei Q (November 2002). "Rapid assessment of repair of ultraviolet DNA damage with a modified host-cell reactivation assay using a luciferase reporter gene and correlation with polymorphisms of DNA repair genes in normal human lymphocytes". Mutation Research. 509 (1–2): 165–174. doi:10.1016/S0027-5107(02)00219-1. PMID 12427537.
- Huang MY, Fang WY, Lee SC, Cheng TL, Wang JY, Lin SR (2008). "ERCC2 2251A>C genetic polymorphism was highly correlated with early relapse in high-risk stage II and stage III colorectal cancer patients: a preliminary study". BMC Cancer. 8: 50. doi:10.1186/1471-2407-8-50. PMC 2262891. PMID 18267032.
- Spitz MR, Wu X, Wang Y, Wang LE, Shete S, Amos CI, Guo Z, Lei L, Mohrenweiser H, Wei Q (February 2001). "Modulation of nucleotide excision repair capacity by XPD polymorphisms in lung cancer patients". Cancer Research. 61 (4): 1354–1357. PMID 11245433.
- Vermeij WP, Dollé ME, Reiling E, Jaarsma D, Payan-Gomez C, Bombardieri CR, Wu H, Roks AJ, Botter SM, van der Eerden BC, Youssef SA, Kuiper RV, Nagarajah B, van Oostrom CT, Brandt RM, Barnhoorn S, Imholz S, Pennings JL, de Bruin A, Gyenis Á, Pothof J, Vijg J, van Steeg H, Hoeijmakers JH (2016). "Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice". Nature. 537 (7620): 427–431. Bibcode:2016Natur.537..427V. doi:10.1038/nature19329. PMC 5161687. PMID 27556946.
- Andressoo JO, Hoeijmakers JH, Mitchell JR (2006). "Nucleotide excision repair disorders and the balance between cancer and aging". Cell Cycle. 5 (24): 2886–8. doi:10.4161/cc.5.24.3565. PMID 17172862.
- Fuss JO, Tainer JA (2011). "XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase". DNA Repair (Amst.). 10 (7): 697–713. doi:10.1016/j.dnarep.2011.04.028. PMC 3234290. PMID 21571596.
- Andressoo JO, Mitchell JR, de Wit J, Hoogstraten D, Volker M, Toussaint W, Speksnijder E, Beems RB, van Steeg H, Jans J, de Zeeuw CI, Jaspers NG, Raams A, Lehmann AR, Vermeulen W, Hoeijmakers JH, van der Horst GT (2006). "An Xpd mouse model for the combined xeroderma pigmentosum/Cockayne syndrome exhibiting both cancer predisposition and segmental progeria". Cancer Cell. 10 (2): 121–32. doi:10.1016/j.ccr.2006.05.027. PMID 16904611.
- Oh KS, Khan SG, Jaspers NG, Raams A, Ueda T, Lehmann A, Friedmann PS, Emmert S, Gratchev A, Lachlan K, Lucassan A, Baker CC, Kraemer KH (2006). "Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome". Hum. Mutat. 27 (11): 1092–103. doi:10.1002/humu.20392. PMID 16947863. S2CID 22852219.
- Gregg SQ, Robinson AR, Niedernhofer LJ (2011). "Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease". DNA Repair (Amst.). 10 (7): 781–91. doi:10.1016/j.dnarep.2011.04.026. PMC 3139823. PMID 21612988.
- Barnhoorn S, Uittenboogaard LM, Jaarsma D, Vermeij WP, Tresini M, Weymaere M, Menoni H, Brandt RM, de Waard MC, Botter SM, Sarker AH, Jaspers NG, van der Horst GT, Cooper PK, Hoeijmakers JH, van der Pluijm I (2014). "Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency". PLOS Genet. 10 (10): e1004686. doi:10.1371/journal.pgen.1004686. PMC 4191938. PMID 25299392.
- Iyama T, Wilson DM (2016). "Elements That Regulate the DNA Damage Response of Proteins Defective in Cockayne Syndrome". J. Mol. Biol. 428 (1): 62–78. doi:10.1016/j.jmb.2015.11.020. PMC 4738086. PMID 26616585.
- Koch S, Garcia Gonzalez O, Assfalg R, Schelling A, Schäfer P, Scharffetter-Kochanek K, Iben S (2014). "Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth". Cell Cycle. 13 (13): 2029–37. doi:10.4161/cc.29018. PMC 4111694. PMID 24781187.
- Edifizi D, Schumacher B (2015). "Genome Instability in Development and Aging: Insights from Nucleotide Excision Repair in Humans, Mice, and Worms". Biomolecules. 5 (3): 1855–69. doi:10.3390/biom5031855. PMC 4598778. PMID 26287260.
- Gorbunova V, Seluanov A, Mao Z, Hine C (2007). "Changes in DNA repair during aging". Nucleic Acids Res. 35 (22): 7466–74. doi:10.1093/nar/gkm756. PMC 2190694. PMID 17913742.
- Goukassian D, Gad F, Yaar M, Eller MS, Nehal US, Gilchrest BA (2000). "Mechanisms and implications of the age-associated decrease in DNA repair capacity". FASEB J. 14 (10): 1325–34. doi:10.1096/fj.14.10.1325. PMID 10877825.
Further reading
- Ellenberger T, Friedberg EC, Walker GS, Wolfram S, Wood RJ, Schultz R (2006). DNA repair and mutagenesis. Washington, D.C: ASM Press. ISBN 978-1-55581-319-2.
- Satoh MS, Hanawalt PC (September 1996). "TFIIH-mediated nucleotide excision repair and initiation of mRNA transcription in an optimized cell-free DNA repair and RNA transcription assay". Nucleic Acids Research. 24 (18): 3576–3582. doi:10.1093/nar/24.18.3576. PMC 146147. PMID 8836185. Article on the relation between TFIIH and NER
- Frit P, Kwon K, Coin F, Auriol J, Dubaele S, Salles B, Egly JM (December 2002). "Transcriptional activators stimulate DNA repair". Mol. Cell. 10 (6): 1391–1401. doi:10.1016/S1097-2765(02)00732-3. PMID 12504014.
- Mellon I (September 2005). "Transcription-coupled repair: a complex affair". Mutat. Res. 577 (1–2): 155–161. doi:10.1016/j.mrfmmm.2005.03.016. PMID 15913669.
External links
 Media related to Nucleotide excision repair at Wikimedia Commons
Media related to Nucleotide excision repair at Wikimedia Commons
| Excision repair | |
|---|---|
| Other forms of repair | |
| Other/ungrouped proteins | |
| Regulation | |
| Other/ungrouped | |
| |