Linnett double-quartet theory
Linnett double-quartet theory (LDQ) is a method of describing the bonding in molecules which involves separating the electrons depending on their spin, placing them into separate 'spin tetrahedra' to minimise the Pauli repulsions between electrons of the same spin. Introduced by J. W. Linnett in his 1961 monograph[1] and 1964 book,[2] this method expands on the electron dot structures pioneered by G. N. Lewis. While the theory retains the requirement for fulfilling the octet rule, it dispenses with the need to force electrons into coincident pairs. Instead, the theory stipulates that the four electrons of a given spin should maximise the distances between each other, resulting in a net tetrahedral electronic arrangement that is the fundamental molecular building block of the theory.

By taking cognisance of both the charge and the spin of the electrons, the theory can describe bonding situations beyond those invoking electron pairs, for example two-centre one-electron bonds. This approach thus facilitates the generation of molecular structures which accurately reflect the physical properties of the corresponding molecules, for example molecular oxygen, benzene, nitric oxide or diborane. Additionally, the method has enjoyed some success for generating the molecular structures of excited states, radicals, and reaction intermediates. The theory has also facilitated a more complete understanding of chemical reactivity, hypervalent bonding and three-centre bonding.
Historical background
The cornerstone of classical bonding theories is the Lewis structure, published by G. N. Lewis in 1916 and continuing to be widely taught and disseminated to this day.[3] In this theory, the electrons in bonds are believed to pair up, forming electron pairs which result in the binding of nuclei. While Lewis’ model could explain the structures of many molecules, Lewis himself could not rationalise why electrons, negatively-charged particles which should repel, were able to form electron pairs in molecules or even why electrons can form a bond between atoms.[4]
Lewis’ theory has been seminal in the understanding of the chemical bond. Yet despite this, it was formulated before the discovery of electron spin, a key intrinsic property of electrons which manifests itself through inter-electronic interactions. While spin was known about ever since the publication of Stern and Gerlach's results in 1922, with the Pauli exclusion principle being formulated in 1925, the importance of 'spin correlation' for understanding when and why electrons form pairs in molecules was not understood until the work of Lennard-Jones in the 1950s.[5] During the latter decade, J. W. Linnett and his students began to explicitly study the role of spin in determining the electronic structures of various molecules.[6][7] This resulted in Linnett's landmark 1961 publication,[1] and subsequent 1964 book,[2] in which he outlined what became known as “Linnett double-quartet” theory.
Linnett continued to expand on his theory through a number of publications[8][9][10][11][12] until his death in 1975. In these writings, Linnett recognised the continued importance of the Lewis model of bonding and the importance of satisfying the octet rule. However, he also argued that this view overemphasises the importance of electron pairing in the formation of chemical bonds. Hence, his theory sought to introduce spin into the conventional model of bonding and hence rectify some of the problems associated with Lewis’ theory. While LDQ theory is a relatively simple extension of Lewis’ bonding theory, the additional freedom of the electrons to separate into two sets, differentiated by their spins, has bestowed upon the theory exquisite agreement with the results of many experiments.[13][14][15][16][17]
In its nascent years, LDQ theory attracted the interest of many researchers, furnishing greater insights into the structures of many molecules. However, LDQ theory began to fade from the spotlight in the 1970s and was mostly abandoned by researchers in the United States, Great Britain and Europe by the mid-1980s.[18]
Formulation of Linnett double-quartet theory
Basic principles
A key trait of LDQ theory that is shared with Lewis theory is the importance of using formal charges to determine the most important electronic structure.[19] LDQ theory produces the spatial distributions of the electrons by considering the two fundamental physical properties of said electrons:
- The mutual repulsion of electrons with like spins, in accordance with the Pauli exclusion principle. Hence, electrons with like (parallel) spins tend to keep as far away from each other as possible by refusing to occupy the same spatial region, while electrons with unlike (antiparallel) spins can occupy the same spatial region. This effect is known as ‘spin correlation’.
- The mutual Coulombic repulsion between electrons. This effect tends to keep electrons as far away from each other as possible, regardless of their relative spins. This is known as ‘charge correlation’.
In Linnett's interpretation, correlation is “the mutual effect the electrons have on one another’s spatial positions”.[7] In the absence of charge correlation, the situation would be as follows:
- If an equal number of both spins is present, the electrons will tend to pair up.
- If an unequal number of spins is present, then the probability distribution of the possible structures is independent of the mutual disposition of the two spin sets.
When one adds the effects of charge correlation, the situation is modified somewhat:
- For electrons with the same spin, charge correlation works in tandem with spin correlation to yield a strong repulsion between the electrons.
- For electrons of opposite spin, charge correlation effects will work against spin correlation effects.
Given these rules, it is found that:
- The four electrons in the same spin set will always keep apart as they experience a negative charge correlation and a negative spin correlation.
- Electrons in different spin sets can pair up (occupy the same spatial region) as they experience a negative charge correlation (which tends to keep them apart) but a positive spin correlation (which favours the spatial proximity of electrons with unlike spins).
Consequences of electron correlation effects
An octet is any arrangement which results in a given nucleus having a total of eight valence electrons around it. In Lewis' bonding model, the electrons tend to pair up in bonds such that an atom has a total of four chemical bonds and lone pairs associated with it: thus, the atom can satisfy its octet. LDQ theory also acknowledges that the elements in the ‘first short period’ of the periodic table tend to attain an octet of electrons surrounding them. However, in contrast with Lewis' view, Linnett argued that due to the combined effects of charge correlation and spin correlation, it is physically more meaningful to consider the octet as the sum of two tetrahedral quartets of electrons. Each quartet consists of electrons of one spin only, and these electrons can act and orient themselves independently. One can then obtain molecular structures by arranging the electrons in such a way as to maximise the separations between the electrons, hence minimising the mutual inter-electronic repulsions, while simultaneously ensuring that the basic geometry of the spin sets is not altered. Additionally, Linnett stressed that due to the Pauli exclusion principle, one should prioritise separating electrons of the same spin when considering the overall electronic structure.
Influence of nearby nuclei

In chemical bonding, the presence of additional nuclei causes the electrons to seek to maximise their attractive electrostatic interactions with all nearby nuclei. This can result in the formation of coincident or ‘close-paired’ electron pairs, in accordance with Lewis’ bonding model. Thus, it has previously[20] been argued that the following should also be included in the basic postulates of LDQ theory:
- The attraction between the nuclei and the electrons tends to distort the electronic geometry. This distortion acts to force a maximum number of electrons into the internuclear (bond axis) region, helping to efficiently bind the nuclei together.
- The presence of any additional nearby nuclei can partially relax the influence of correlation effects on the electronic geometry. Therefore, it is possible for two electrons of opposite spin to come together and occupy the same spatial region, effectively forming the classical Lewis electron pair. This can serve to strengthen the binding between the nuclei by increasing the net electron density in the internuclear region. The exact disposition of the electrons is determined by the relative electronegativities of the constituent elements.
The electron pairing can result in a greater net binding between the nuclei, but this is not necessarily the case in all molecules. In his discussions,[1][2] Linnett notes that due to the opposing effects of charge and spin, the correlation between the two spin quartets should be small and so the individual spin tetrahedra can be treated as being partly independent from each other. This then facilitates electron pairing since nearby nuclei can easily force the two electrons together. Linnett also argues that a relatively small deviation from the strictly regular tetrahedra of the rigorous LDQ theory approach could be energetically favourable in some cases.
Balancing the intramolecular interactions
The structure obtained from applying LDQ theory balances the three principal interactions in the molecule: electron-electron, electron-nuclear and nuclear-nuclear. Much like Lewis’ bonding model, LDQ theory assumes that the dominant contributions result from electron-electron and electron-nuclear interactions.[20] However, it has previously been shown that the introduction of nuclear-nuclear interactions into LDQ theory can explain some trends in bond angles and bond lengths.[20] In particular, Firestone produced an extensive discussion[21][22] of the effects of moving bonding electron density out of the internuclear region and highlighted that sometimes such a distortion is necessary to produce a more satisfactory arrangement of the spin sets. Due to the decreased shielding of the nuclear-nuclear interactions and the decreased electron-nuclear interactions associated with this change, the net energy of the molecule tends to increase: this is known as “L-strain” (see section on reactivity later).
Examples of the application of the theory
Understanding structures using LDQ
As an example of the application of LDQ theory to molecular bonding, take the case of the fluoride ion. By using LDQ theory, the electronic structure shown below is obtained.
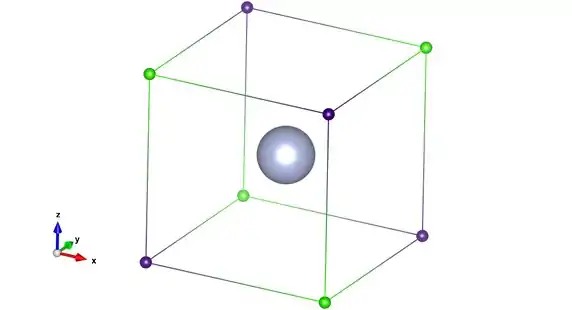
The two spin sets are under the action of only one nucleus and so there is no net interaction which will cause the electrons to pair up. Hence, unlike the Lewis model which predicts four lone pairs, all electrons in the fluoride ion are spatially separated. Therefore, the following statement by Luder is found to be true for all mononuclear species:[23]
“In an isolated atom, no valence electron is close-paired with another”.
If a proton then approaches the fluoride ion, the proton's attractive potential can distort the electronic geometry. Two electrons of opposite spin (necessary to complete the duplet of the hydrogen atom) are attracted to the proton and this attractive potential pulls them together to yield an electron pair localised to the internuclear region. This is illustrated in the LDQ structure of hydrogen fluoride shown below.

Again, while the Lewis picture would predict four coincident electron pairs, the LDQ theory treatment yields only one close pair and two staggered spin tetrahedra that share a vertex. This makes sense as the other six electrons, unlike the two bonding electrons, do not significantly experience the attractive influence of the proton and hence their inter-electronic repulsions keep them separated.
Example: molecular oxygen
One of the major triumphs of LDQ theory over the traditional Lewis view is the ability of the former to generate an electronic structure which explains the paramagnetism of the ground state (3Σg− state) of molecular oxygen (O2). The LDQ structure of the ground state of O2 does not involve any electron pairs, in contrast with the Lewis structure of the molecule. Instead, the electrons are arranged as shown below.

There are seven valence electrons of one spin which occupy two tetrahedra that share a common vertex (purple spheres), and the remaining five valence electrons of the other spin occupy two tetrahedra which share a common face (green spheres). Linnett postulated that this electronic arrangement reduces the magnitude of the inter-electronic repulsions in comparison with the case where the two spin sets have six electrons each. This arrangement results in a bond order of 2 and an excess of one electron spin, giving rise to the molecule's paramagnetism: both observations are in agreement with molecular orbital theory treatments of the molecule. In effect, the LDQ structure is equivalent to the combination of a two-centre one-electron bond (purple spin set) and a two-centre three-electron bond (green spin set).
Example: methane
Not all LDQ structures differ from those produced using Lewis’ bonding model. For example, an alkane such as methane has both spin tetrahedra totally coincident, resulting in four close-pairs of electrons as in the Lewis picture.

Simplification of the theory: 2D structures
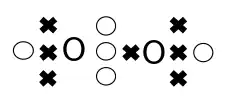
The above three-dimensional LDQ structures are useful for visualising the molecular structures, but they can be laborious to construct. Hence, Linnett introduced two-dimensional structures, analogous to Lewis structures, that used dots and crosses to represent the relative spin states of electrons. An example is shown on the right for molecular oxygen.
Further, Linnett also modified the lines used in Lewis structures to account for electron coincidence and/or non-coincidence: a thin line represents an electron pair that is not close-paired, while a thick line represents a close-pair of electrons. This is exemplified best in the case of the hydrogen fluoride molecule, the dot-and-cross diagram of which is shown on the right. Here, the Lewis structure drawn on the left of the image is compared with the LDQ line structure on the right of the image. The LDQ structure thus expands on the Lewis structure by denoting if the electrons are coincident (thick line) or if they are spatially separated (thin lines).

Additionally, by adding a dot or cross above/below the bond line, one can denote an odd number of electrons which are involved in the bond. This is illustrated well in the structure of nitric oxide (NO) shown below:

More details about the LDQ structures of radicals such as NO are given in the section ‘Theoretical Description of Radicals’.
Example: benzene
LDQ theory has been lauded for its ability to produce an accurate electronic structure of benzene.[18][19] The LDQ structure for benzene is shown below.[16][24]


In this model, each carbon atom is bonded to its neighbouring carbon atoms by three non-coincident electrons, two of one spin (e.g. green spheres) and one of the other spin (e.g. purple spheres). Thus, LDQ theory is able to predict the 1.5 bond order of the carbon-carbon bonds in benzene, the equivalence of all six carbon-carbon bonds and the stability of benzene due to the fact that none of the electrons in the carbon-carbon bonds are close-paired. This is in contrast with the valence bond picture which must invoke resonance between the two Kekulé forms of benzene in order to predict the non-integral bond order. Hence, the LDQ structure is lower in energy than either of the Kekulé forms due to a reduction in the magnitude of the inter-electronic repulsions in the former.
The 2D LDQ structures of benzene using both the full dot-and-cross diagram and the simplified diagram are shown on the right. Again, the bonding situation determined using LDQ theory is in good agreement with molecular orbital theory results.[2][18] This also highlights that the additional degree of freedom afforded by having two distinct spin sets in the LDQ approach allows a single electron in a bond to be shared equally between two atoms, which produces the above structure for benzene.
Theoretical description of excited states
The ability of LDQ theory to describe electronic distributions in terms of independent spin sets has facilitated studies of the excited states of various molecules, producing excited state electronic structures that are in agreement with experiments.[2] This sets LDQ theory apart from both valence bond theory and Lewis bonding theory as these have not been previously utilised to study excited state electronic structures.[18] Further, the LDQ theory approach to studying excited states produces three-dimensional redistributions of the electron density, in contrast with the single-electron vertical transitions produced using molecular orbital methods.
Example: excited states of molecular oxygen
As outlined previously, Linnett found that disposing the electrons into two spin sets, one with seven electrons and the other with only five electrons, produced the electronic structure of the ground state of O2 (see above).[2] In contrast, one can look at the case where the two spin sets both contain six electrons to generate the excited states of O2. When the spin sets are non-coincident, the electronic structure shown below is produced.

In this case, each spin set is the same but there is no correlation between them, giving rise to a cubic arrangement of the electrons. As the average distance between the electrons is shorter than in the ground state case, this disposition of the electrons thus results in a greater net magnitude of the inter-electronic repulsion energy as compared to the ground state. Hence, the above structure corresponds to the first excited state (1Δg state) of O2. If one further increases the degree of inter-electronic repulsions by forcing the electrons into coincident pairs, the electronic structure shown below is generated.[2]

This corresponds to the electronic structure of the second excited state of O2 (1Σg+ state), and also corresponds to the (incorrect) Lewis structure of the ground state of O2. Thus, a comparison of the magnitude of the inter-electronic repulsions in a series of possible molecular structures can be used to assess their relative energies and hence determine the ground and excited states.
Additionally, it is found that in all three electronic structures, the net bond order is 2 as they all have four electrons in the spatial region between the oxygen nuclei. Thus, we see that this example clearly demonstrates that “not all double bonds are created equal”.[18]
Example: excited states of acetylene
Linnett also used the example of acetylene to illustrate the power of the LDQ approach for understanding the structures of the excited states of molecules.[2] The dot-and-cross diagrams for both the ground state and the first excited state of acetylene are shown below.

Upon excitation of the acetylene molecule, there is a net depletion of electron density from the bond region. This is captured in the above figure on the right as three electrons are withdrawn from the internuclear region and localised to the individual carbon atoms: resonance needs to be invoked in this case to explain how the three electrons can be distributed among the two carbon centres. Linnett rationalises this three-electron redistribution by arguing that it is required by the need to both form the two carbon-hydrogen bonds and retain the tetrahedral disposition of the electrons of a given spin.[2][25] Interestingly, the excited state does not obey the octet rule as the carbon atoms have an average 6.5 valence electrons surrounding them. Further, the internuclear region contains only three electrons, the same as in the benzene molecule (see above), and this explains why the carbon-carbon bond length in the excited state of acetylene is the same as that in benzene.[1][25] Most strikingly, the molecule changes its geometry upon excitation, going from a simple linear symmetry to a trans-bent structure. This is in excellent agreement with both the landmark results of Ingold and King,[26] which were the first demonstration of an excited state having a qualitatively different geometry than the ground state, and the results from molecular orbital theory methods.[25] Thus, this example illustrates that LDQ theory can be a powerful tool for understanding the geometric rearrangements that occur when excited states are formed.
Theoretical description of radicals
A major drawback of Lewis’ bonding theory is its inability to predict and understand the structures of radicals due to the presence of unpaired single electrons. LDQ theory has seen great success in explaining the structures of open shell systems such as nitric oxide or ozone due to the additional degree of freedom associated with having two independent spin sets. In the cases of nitric oxide and ozone, the maxima of the electron density of the localised orbitals result in distributions which closely mirror the dot-and-cross diagrams produced using LDQ theory.[27]
Example: nitric oxide
The typical example of a radical that cannot be treated satisfactorily using Lewis structures is nitric oxide (NO). By allowing the electrons in the two spin sets to separate from each other, the LDQ structure for NO can be generated as shown below.

Hence, the NO molecule is held together by a perfectly symmetric two-centre five-electron bond, made up of three electrons of one spin (green spheres) and two electrons of the other spin (purple spheres). This bonding arrangement satisfies the octet for both the nitrogen and oxygen atoms and results in a bond order of 2.5, in excellent agreement with the molecular orbital theory treatment of NO.[2]
Stability of radicals against dimerisation
It has previously been highlighted that, from applications of LDQ theory, there exist two distinct classes of radicals: (a) radicals which do not have enough electrons to satisfy the octets of their constituent atoms and (b) radicals which obey the octet rule. Radicals of type (a) are thus highly reactive fragments which want to gain electrons to satisfy the octet rule, while radicals of type (b) are stable species by virtue of satisfying the octets of their constituent atoms.[18]
As an example, the cyanide (CN) radical shown below is a type (a) radical that has ten bonding electrons, while the cyanogen molecule (a dimeric combination of two CN radicals) has 14 bonding electrons.
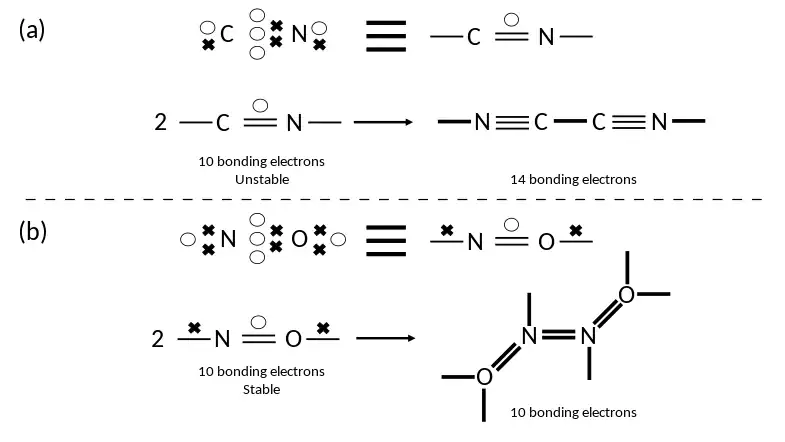
Hence, the dimerisation of CN to cyanogen is favourable as it increases the degree of bonding in the overall system and reduces the total energy. In contrast, the NO molecule is a type (b) radical, also with ten electrons. However, the dimeric N2O2 molecule likewise has ten bonding electrons, and hence there is no significant energetic benefit from the formation of the dimer. In fact, the formation of the nitrogen-nitrogen bond leads to an increase in the number of close-paired electrons and hence an increase in the total system energy, and so isolated NO molecules are stable against dimerisation in the gas phase.[2]
Application to chemical reactivity
LDQ theory has enjoyed some success in studies of chemical reactivity, in particular organic reactions, as it can furnish one with the ability to predict chemical reactivity from analyses of the relevant reactant and transition state structures. Firestone's extensive work constitutes the most significant application of LDQ theory to chemical reactivity thus far.[21][22][28][29][30][31][32][33] Firestone has previously used the concept of L-strain (see above) to analyse the activation energies in SN2, SH2 and E2 reactions, since the movement of electron density out of the internuclear region is commonly associated with the formation of transition states.
Example: reactivity among different families of hydrocarbons
LDQ structures, in particular the coincidence of electron pairs, can be used to rationalise and explain the stability and reactivity of certain families of molecules such as hydrocarbons.

As shown for ethane, the electrons reside in two coincident tetrahedra which share a common vertex, and hence all the electrons are in close-pairs as expected from Lewis’ bonding model. However, compare this with the situation in ethylene: again, all the electrons are in close-pairs but now there is no electron density along the internuclear axis. The result is that the energy required to overcome charge correlation and pair the electrons up is compensated to a lesser extent by the bonding in ethylene as compared with ethane. Thus, in agreement with experiments, the ethylene molecule should be highly reactive with respect to addition reactions. Finally, the above can be compared with the situation in acetylene. Here, the six electrons involved in bonding are all anti-coincident and so the energy cost associated with charge correlation is minimised. Indeed, in agreement with experiment, carbon-carbon triple bonds are far less reactive with respect to addition reactions than carbon-carbon double bonds as transforming carbon-carbon triple bonds into double bonds also involves the formation of close-pairs of electrons, an energetically costly process.[17][34]
Application to hypervalent and three-centre bonding
The strengths of LDQ theory have been applied to understand the structures and bonding modes of various molecules which, in the valence bond method, are described using the terms ‘hypervalent’ and ‘three-centre bonding’.
Hypervalent molecules

In the case of phosphorus pentachloride (PCl5), the example shown on the right, the central phosphorus atom is bonded to five chlorine atoms. In the traditional Lewis view, this violates the octet rule as the five phosphorus-chlorine bonds would result in a net ten electrons around the phosphorus atom. Thus, the molecule is assumed to expand its bonding beyond the octet, a situation known as hypervalent bonding.
LDQ theory, however, presents a different view of the bonding in this molecule. The three equatorial chlorine atoms each form two-electron bonds with the central phosphorus atom. The remaining two axial chlorine atoms each contribute only one electron to a bond with the phosphorus atom, leaving a single electron to reside exclusively on the chlorine atom. Thus, the LDQ structure for PCl5 consists of three two-centre two-electron bonds and two two-centre one-electron bonds, thus satisfying the octet rule and dispensing with the need to invoke hypervalent bonding. This LDQ structure is also in good agreement with quantum chemical calculations.[18]
Three-centre bonding
LDQ theory has facilitated a more rigorous analysis of bonding in compounds which have conventionally been described in terms of three-centre two-electron bonding. For example, compare the various ways shown below to represent the bonding in the Lewis acid-base adduct of the hydride anion (H−) and borane (BH3) shown below.

The LDQ approach thus enables each electron to localise in one of the boron-hydrogen internuclear bond regions, rather than being delocalised over the entire three-centre boron-hydrogen-boron moiety. This arrangement of the bonding electrons into two two-centre one-electron bonds benefits from a lowering of the net magnitude of the inter-electronic repulsions in the system. In comparison, as described by Linnett:[2]
“By allowing the two electrons independent ‘movement’ in a three-centre system, the three-centre bond allows the electrons a fairly considerable chance of being near one another”.
Similarly, the resonance forms shown above also increase the degree of inter-electronic repulsions as the electrons are paired up in the boron-hydrogen bonds. Thus, a more complete description of the bonding in B2H7− is obtained using LDQ theory as it can utilise two two-centre one-electron bonds, in comparison with the awkward three-centre two-electron bond or the resonance structures derived from the valence bond method.
The situation is similar for diborane (B2H6), the archetypal example used to explain three-centre two-electron bonding.[35]

The above demonstrates that the structure produced using LDQ theory again yields the lowest degree of inter-electronic repulsions.
Indeed, the separation of the electrons into two distinct spin sets has enabled the theory to expand the set of possible bonding arrangements, with two-centre one-electron, two-centre three-electron and two-centre five-electron bonding patterns all possible in the theory.[18]
Quantitative extension of Linnett double-quartet theory
Along with the qualitative picture outlined above, LDQ theory has also been applied to computational studies. This quantitative extension is known as the non-pairing spatial orbital (NPSO) theory. In the NPSO method, the constituent wave functions are based on the corresponding qualitative LDQ structures. This approach has previously been shown to produce lower energies as compared to valence bond or molecular orbital wave functions derived from Lewis structures for molecules such as benzene, diborane or ozone.[36][37][16][15][13][38][39] Hence, by the variational principle, the wave functions produced by NPSO methods are often a better approximation than those generated using molecular orbital theory methods.
Relation to the electron localisation function
It is possible to visualise the reality of disposing the two spin sets separately. Recent investigations have shown that the electron localisation function (ELF) can be successfully applied to understand the disposition of the electrons in a number of molecules.
Example: acetylene
The ELF of acetylene has been studied by a number of authors.[4][40][41] The results of this analysis are indicated in the figure below.

The ELF of acetylene thus contains a toroidal basin surrounding the carbon-carbon bond axis, rather than three discrete concentrations of electron density as would be expected from the Lewis structure for a triple bond. This is directly comparable to the bonding picture produced using LDQ theory (see above), highlighting that the theory can accurately reflect the bonding situation in multiply-bonded species.[4]
Example: digermyne
A recent report[42] on the disilyne and digermyne molecules has shown that their ELFs also result in a toroidal basin surrounding the internuclear axis.

The toroidal basin represents the six electrons which are involved in the bonding between the two germanium centres in this molecule. The LDQ structure is in excellent agreement with these computational results: the toroid is angled in comparison with the case in acetylene due to the perturbation caused by the off-axis hydrogen atoms.
Example: chlorine trifluoride
In the VSEPR structure of chlorine trifluoride (ClF3), the molecule adopts a trigonal bipyramidal structure with the central chlorine atom violating the octet rule. This is typically rationalised by invoking d orbital participation in the bonding of the sp3d hybridised chlorine centre.[43] The ELF of ClF3 is presented below.

The ELF analysis of ClF3 indicates that there is a single toroidal-shaped basin at the 'back' of each fluorine atom, corresponding analogously to the three lone pairs arranged in a ring as generated for the HF molecule (see above). This is in contrast with the Lewis structure which would place the fluorine lone pair electrons into discrete coincident pairs. Further, the lone pairs of electrons associated with the central chlorine atom reside in two kidney-shaped lobes which lie in the equatorial plane along with one of the fluorine atoms. This structure, consistent with the LDQ structure of the molecule, is also consistent with the VSEPR structure as the more diffuse chlorine lone pairs distort the molecular geometry and result in the bent planar geometry seen.[43][44] In contrast, the bonding situation described by LDQ theory differs greatly from that produced using valence bond theory. Rather than having three two-centre two-electron bonds and two lone pairs, necessitating the invocation of hypervalent bonding for the chlorine atom, the LDQ structure instead allows the axial fluorine atoms to form two-centre one-electron bonds. This, when combined with a two-centre two-electron bond to the equatorial fluorine atom and the two chlorine lone pairs, restores the octet of the chlorine atom. As exemplified by the increased bond length of the axial fluorine-chlorine bonds as compared to the equatorial fluorine-chlorine bond,[45] LDQ theory is able to more accurately describe the electronic structure of ClF3 as compared to valence bond theory.
Strengths and weaknesses of the theory
Strengths of the approach
One of the main benefits is that many molecular structures, such as molecular oxygen and ozone, can be represented using a single LDQ structure without invoking any resonance structures. This lesser reliance on resonance structures is favourable as, according to Linnett, resonance structures are not satisfactory descriptions of bonding as the ‘resonance stabilisation energy’ is not easily attributable to any particular molecular feature.[2]
Several other strengths of the approach include:
- It can be used to generate the electronic structures of species with π systems, affording greater precision for systems where there are partial charges associated with the constituent atoms.[46]
- It is able to treat individual molecular features, such as π systems, separately from the rest of the structure. This is in contrast with molecular orbital theory approaches which often require the simultaneous treatment of σ and π systems.
- It can be used to understand and predict the relative bond strengths of a single species in cases where a number of structures with different bond orders are possible.
The success of LDQ theory in elucidating structures akin to those generated using quantum chemical calculations has also afforded a better understanding of the meaning of the dots and crosses used in the theory. Accordingly, the dots and crosses have been associated with the centroids of charge of the localised orbitals, while also making the distinction between the two sets of spins in the charge analyses.[19][47]
Weaknesses of the approach
LDQ theory greatly diminishes, but does not completely remove, the need for invoking resonance structures to explain the bonding in certain molecules. While the need for resonance structures is reduced, it is still necessary to invoke resonance for certain molecules such as semiquinones, nitryl chloride or nitrogen dioxide.
Additionally, like its Lewis theory progenitor, the theory ignores the energy differences between s and p orbitals. This has garnered criticism from authors who have dismissed LDQ theory as it was seen to invoke "the inert gas magic".[48] Other authors have also claimed that LDQ theory cannot be easily extended "to larger systems for which its use generally becomes very intuitive" and that its results are "as ambiguous as those of resonance theory".[49]
Luder’s extension of Linnett double-quartet theory – electron-repulsion theory
Linnett's vision of double-quartet theory was limited to elements which did not expand their valence beyond the octet: this produced the familiar spin tetrahedra. However, later work by W. F. Luder[23][50] extended the principles of LDQ theory to produce electronic structures with more than four electrons in each spin set. This extension, called “electron repulsion theory” by Luder, could be applied to elements of the d and f blocks in the periodic table.

For example, the structure of the zinc atom produced using electron-repulsion theory is shown above. The author asserts that the s electrons occupy the axial positions, leaving the d electrons to occupy the positions at the vertices of two pentagonal bases of the two constituent pyramids. The electronic structure of the ytterbium atom can be constructed similarly. The s electrons are again assumed to occupy the axial positions while the f electrons occupy the positions at the vertices of two heptagonal bases of the two constituent pyramids.
While these results are interesting, they have been contested in the scientific literature due to Luder's abandonment of the octet rule and the author's controversial views on spin correlation.[18] Indeed, one author notes that Luder's works “[do] a great disservice to Linnett and his method”.[51]
Recent applications of Linnett double-quartet theory
Recently, there has been a modest resurgence of LDQ theory in the scientific literature, especially among theoretical chemists. For example, a recent study found that there is a qualitative correspondence between the molecular structures produced using LDQ theory and those suggested by dynamic Voronoi metropolis sampling.[52]
Another recent example is the correspondence of the results obtained using LDQ theory to those produced using the Fermi-Löwdin orbital self-interaction correction[53] (FLO-SIC) method. It was shown that this method generates structures which can successfully house two electrons of one spin in a given ‘spin channel’, and the remaining single electron can be housed in the other spin channel:[54] this can be directly related to the LDQ structures of many radicals (see for instance NO above). Further, the electronic geometries for many ground state molecules, such as carbon dioxide, produced via FLO-SIC methods were found to generally agree with those derived from LDQ theory. In a subsequent publication, the authors posited that the Fermi orbital descriptors[55] utilised in their work can be correlated to the electron spins generated in LDQ analyses.[56] The authors also noted that the use of LDQ theory to produce model electronic structures of molecules for quantum calculations results in calculated dipole moments that agree more closely with experiments.
References
- Linnett, J. W. (June 1961). "A Modification of the Lewis-Langmuir Octet Rule". Journal of the American Chemical Society. 83 (12): 2643–2653. doi:10.1021/ja01473a011. ISSN 0002-7863.
- Linnett, John Wilfred (1964). The Electronic Structure of Molecules: A New Approach. London: Methuen.
- Lewis, Gilbert N. (1916-04-01). "The Atom and the Molecule". Journal of the American Chemical Society. 38 (4): 762–785. doi:10.1021/ja02261a002. ISSN 0002-7863. S2CID 95865413.
- Gillespie, R. J.; Robinson, E. A. (2007-01-15). "Gilbert N. Lewis and the chemical bond: The electron pair and the octet rule from 1916 to the present day". Journal of Computational Chemistry. 28 (1): 87–97. doi:10.1002/jcc.20545. PMID 17109437. S2CID 186530.
- Lennard‐Jones, J. E. (1952-06-01). "The Spatial Correlation of Electrons in Molecules". The Journal of Chemical Physics. 20 (6): 1024–1029. Bibcode:1952JChPh..20.1024L. doi:10.1063/1.1700620. ISSN 0021-9606.
- Linnett, J. W.; Poë, A. J. (1951). "Directed valency in elements of the first short period". Trans. Faraday Soc. 47: 1033–1041. doi:10.1039/TF9514701033. ISSN 0014-7672.
- Mellish, C. E.; Linnett, J. W. (1954). "Directed valency in elements of the first and second short periods". Transactions of the Faraday Society. 50: 657. doi:10.1039/tf9545000657. ISSN 0014-7672.
- LINNETT, J. W. (1964). "Valency and the Chemical Bond". American Scientist. 52 (4): 459–475. ISSN 0003-0996. JSTOR 27839160.
- Linnett, J.W.; Rosenberg, R.M. (1964). "Structure and properties of nitroso compounds". Tetrahedron. 20 (1): 53–66. doi:10.1016/S0040-4020(01)98396-6.
- Linnett, J. W. (1970-03-27). "Structure of Polywater". Science. 167 (3926): 1719–1720. Bibcode:1970Sci...167.1719L. doi:10.1126/science.167.3926.1719. ISSN 0036-8075. PMID 17729618. S2CID 44969568.
- Linnett, J. W. (1971), Downs, A. J.; Long, D. A.; Staveley, L. A. K. (eds.), "Molecular Force Fields and Valency", Essays in Structural Chemistry, Boston, MA: Springer US, pp. 1–17, doi:10.1007/978-1-4684-1902-3_1, ISBN 978-1-4684-1904-7, retrieved 2021-11-17
- Linnett, J. W. (1972). "Chemical bonds". Science Progress (1933- ). 60 (237): 1–23. ISSN 0036-8504. JSTOR 43420121.
- Empedocles, P. B.; Linnett, J. W. (1966). "Ground state wavefunctions of some conjugated carbon compounds - NPSO method". Theoretica Chimica Acta. 4 (4): 377–389. doi:10.1007/BF01129652. ISSN 0040-5744. S2CID 95070064.
- Hirst, D. M.; Linnett, J. W. (1965-11-15). "Electronic Structure of the Nitrite Ion and Related Species". The Journal of Chemical Physics. 43 (10): S74–S79. Bibcode:1965JChPh..43S..74H. doi:10.1063/1.1701516. ISSN 0021-9606.
- Bowen, H. C.; Linnet, J. W. (1964-01-01). "Electronic wave function for H+3, H3, H–3". Transactions of the Faraday Society. 60: 1185–1192. doi:10.1039/TF9646001185. ISSN 0014-7672.
- Empedocles, P. B.; Linnett, John Wilfrid (1964-11-03). "The electronic structure of benzene". Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences. 282 (1389): 166–177. Bibcode:1964RSPSA.282..166E. doi:10.1098/rspa.1964.0224. S2CID 95733000.
- Serratosa, Felix (June 1973). "Linnett's theory and the thermal stability of heterosubstituted acetylenes". Journal of Chemical Education. 50 (6): 402. Bibcode:1973JChEd..50..402S. doi:10.1021/ed050p402. ISSN 0021-9584.
- Jensen, William B. (April 2017). "Whatever happened to Linnett double-quartet (LDQ) theory?". Educación Química. 28 (2): 74–83. doi:10.1016/j.eq.2016.11.003.
- Duke, Brian J. (July 1987). "Linnett's double quartet theory and localised orbitals". Journal of Molecular Structure: THEOCHEM. 152 (3–4): 319–330. doi:10.1016/0166-1280(87)80072-6.
- Jensen, William B. (1981-03-01). "Some comments on Linnett Double Quartet theory". Canadian Journal of Chemistry. 59 (5): 807–813. doi:10.1139/v81-117. ISSN 0008-4042.
- Firestone, Raymond A. (January 1968). "Application of the Linnett electronic theory to organic chemistry". Tetrahedron Letters. 9 (8): 971–975. doi:10.1016/S0040-4039(01)98838-0.
- Firestone, Raymond A. (September 1969). "Application of the Linnett electronic theory to organic chemistry. II. Introduction". The Journal of Organic Chemistry. 34 (9): 2621–2627. doi:10.1021/jo01261a029. ISSN 0022-3263.
- Luder, W. F. (April 1967). "The electron repulsion theory of the chemical bond. I. New models of atomic structure". Journal of Chemical Education. 44 (4): 206. Bibcode:1967JChEd..44..206L. doi:10.1021/ed044p206. ISSN 0021-9584.
- Liu, Yu; Kilby, Phil; Frankcombe, Terry J.; Schmidt, Timothy W. (2020). "The electronic structure of benzene from a tiling of the correlated 126-dimensional wavefunction". Nature Communications. 11 (1): 1210. Bibcode:2020NatCo..11.1210L. doi:10.1038/s41467-020-15039-9. ISSN 2041-1723. PMC 7058002. PMID 32139681.
- Linnett, J. W. (1958-01-01). "Equivalent Orbitals and the Shapes of Excited Species". Canadian Journal of Chemistry. 36 (1): 24–30. doi:10.1139/v58-003. ISSN 0008-4042.
- King, G. W.; Ingold, C. K. (June 1952). "The Bent Excited State of Acetylene". Nature. 169 (4313): 1101–1102. Bibcode:1952Natur.169.1101K. doi:10.1038/1691101b0. ISSN 1476-4687. S2CID 4162767.
- Hirst, D. M.; Linington, Mary E. (1970). "Localized orbitals for the oxygen and nitric oxide molecules". Theoretica Chimica Acta. 16 (1): 55–62. doi:10.1007/BF01045967. ISSN 0040-5744. S2CID 95235964.
- Firestone, Raymond A. (1970-01-01). "Applications of the Linnett electronic theory to organic chemistry. Part III. Linnett structures for 1,3-dipoles and for the diradical intermediates in 1,3-dipolar cycloadditions". Journal of the Chemical Society A: Inorganic, Physical, Theoretical: 1570–1575. doi:10.1039/J19700001570. ISSN 0022-4944.
- Firestone, Raymond A. (March 1971). "Application of the Linnett electronic theory to organic chemistry. IV. SN2 transition state". The Journal of Organic Chemistry. 36 (5): 702–711. doi:10.1021/jo00804a020. ISSN 0022-3263.
- Firestone, Raymond A. (June 1972). "Application of the Linnett electronic theory to organic chemistry. V. Orientation in 1,3-dipolar cycloadditions according to the diradical mechanism. Partial formal charges in the Linnett structures of the diradical intermediate". The Journal of Organic Chemistry. 37 (13): 2181–2191. doi:10.1021/jo00978a027. ISSN 0022-3263.
- Firestone, Raymond A. (1973-01-01). "Linnett planar methane". Journal of the Chemical Society, Chemical Communications (5): 163b–164. doi:10.1039/C3973000163B. ISSN 0022-4936.
- Firestones, Raymond a. (March 1976). "The π-Bond Energy of the Carbonyl Group". Tetrahedron Letters. 17 (10): 735–736. doi:10.1016/S0040-4039(00)77937-8.
- Firestone, Raymond A. (August 1980). "Application of the Linnett electronic theory to organic chemistry. 7. Linnett structures for homolysis transition states. The bridgehead azo anomaly". The Journal of Organic Chemistry. 45 (18): 3604–3609. doi:10.1021/jo01306a012. ISSN 0022-3263.
- Kutepow, Nikolaus von (1972). "Chemistry of acetylenes. Von H. G. Viehe. Marcel Dekker Inc., New York 1969. 1. Aufl., XV, 1298 S., zahlr. Tab. u. Formeln, geb. $ 59.50". Angewandte Chemie. 84 (8): 367. Bibcode:1972AngCh..84..367V. doi:10.1002/ange.19720840843. ISSN 1521-3757.
- Wade, K. (1971), "Structure and bonding of diborane", Electron Deficient Compounds, Boston, MA: Springer US, pp. 6–31, doi:10.1007/978-1-4684-6054-4_2, ISBN 978-1-4684-6056-8, retrieved 2021-11-18
- Hirst, D. M.; Linnett, J. W. (November 1961). A new method for deriving molecular wave functions. Proceedings of the Chemical Society. pp. 427–428. doi:10.1039/PS9610000397.
- Chong, D.P.; Linnett, J.W. (1964-01-01). "Alternant molecular orbital treatment of the allyl cation, radical and anion". Molecular Physics. 8 (2): 151–155. Bibcode:1964MolPh...8..151C. doi:10.1080/00268976400100181. ISSN 0026-8976.
- Gould, R. D.; Linnett, J. W. (1963). "Electronic structure of ozone". Transactions of the Faraday Society. 59: 1001. doi:10.1039/tf9635901001. ISSN 0014-7672.
- Hirst, D. M.; Linnett, J. W. (1962-01-01). "194. Electronic structures and formulœ: the allyl cation, radical, and anion". Journal of the Chemical Society (Resumed): 1035–1047. doi:10.1039/JR9620001035. ISSN 0368-1769.
- Santos, Juan C.; Polo, Victor; Andrés, Juan (2005-05-02). "An electron localization function study of the trimerization of acetylene: Reaction mechanism and development of aromaticity". Chemical Physics Letters. 406 (4): 393–397. Bibcode:2005CPL...406..393S. doi:10.1016/j.cplett.2005.02.102. ISSN 0009-2614.
- Gillespie, R. J. (2008-07-01). "Fifty years of the VSEPR model". Coordination Chemistry Reviews. 252 (12): 1315–1327. doi:10.1016/j.ccr.2007.07.007. ISSN 0010-8545.
- Malcolm, Nathaniel O. J.; Gillespie, Ronald J.; Popelier, Paul L. A. (2002-08-29). "A topological study of homonuclear multiple bonds between the elements of group 14". Journal of the Chemical Society, Dalton Transactions (17): 3333–3341. doi:10.1039/B110610B. ISSN 1364-5447.
- Gropen, O.; Wisløff-Nilssen, E. (1982-08-01). "Electronic structure and bonding in the ClF3 molecule. An extensive RHF—ab initio study". Journal of Molecular Structure: THEOCHEM. 88 (3): 243–248. doi:10.1016/0166-1280(82)80173-5. ISSN 0166-1280.
- Petrucci, Ralph H. (2002). General chemistry : principles and modern applications. William S. Harwood, F. Geoffrey Herring (8th ed.). Upper Saddle River, N.J.: Prentice Hall. pp. 413–414. ISBN 0-13-014329-4. OCLC 46872308.
- Smith, D. F. (1953-04-01). "The Microwave Spectrum and Structure of Chlorine Trifluoride". The Journal of Chemical Physics. 21 (4): 609–614. Bibcode:1953JChPh..21..609S. doi:10.1063/1.1698976. hdl:2027/mdp.39015095092865. ISSN 0021-9606.
- Langler, Richard Francis; Trenholm, June Ellen; Wasson, John Spencer (1980-04-15). "Linnett double quartet theory in organic chemistry: structure, reactivity and resonance". Canadian Journal of Chemistry. 58 (8): 780–785. doi:10.1139/v80-122. ISSN 0008-4042.
- Leroy, Georges (1981), Csizmadia, I. G.; Daudel, R. (eds.), "Structure and Properties of Free-Radicals. A Theoretical Contribution", Computational Theoretical Organic Chemistry, Dordrecht: Springer Netherlands, pp. 253–334, doi:10.1007/978-94-009-8472-1_13, ISBN 978-94-009-8474-5, retrieved 2021-11-18
- Pimentel, George C.; Spratley, Richard D. (1969). Chemical Bonding Clarified Through Quantum Mechanics. Holden-Day. pp. 148–153. ISBN 978-0-8162-6781-1.
- Quantum chemistry. Raymond Daudel. Chichester [West Sussex]: Wiley. 1983. pp. 258–271, 279–283. ISBN 0-471-90135-0. OCLC 9082193.
{{cite book}}: CS1 maint: others (link) - Luder, W. F. (1967). The Electron-repulsion Theory of the Chemical Bond. New York: Reinhold Publishing Corporation.
- Manch, Walter A. (1969-02-01). "The Electron-repulsion theory of the chemical bond. Review 2". Journal of Chemical Education. 46 (2): 126. Bibcode:1969JChEd..46..126M. doi:10.1021/ed046p126.3. ISSN 0021-9584.
- Liu, Yu; Frankcombe, Terry J.; Schmidt, Timothy W. (2016-05-11). "Chemical bonding motifs from a tiling of the many-electron wavefunction". Physical Chemistry Chemical Physics. 18 (19): 13385–13394. Bibcode:2016PCCP...1813385L. doi:10.1039/C6CP01188H. ISSN 1463-9084. PMID 27122062.
- Pederson, Mark R.; Ruzsinszky, Adrienn; Perdew, John P. (2014-03-28). "Communication: Self-interaction correction with unitary invariance in density functional theory". The Journal of Chemical Physics. 140 (12): 121103. Bibcode:2014JChPh.140l1103P. doi:10.1063/1.4869581. ISSN 0021-9606. PMID 24697415.
- Schwalbe, Sebastian; Trepte, Kai; Fiedler, Lenz; Johnson, Alex I.; Kraus, Jakob; Hahn, Torsten; Peralta, Juan E.; Jackson, Koblar A.; Kortus, Jens (2019). "Interpretation and Automatic Generation of Fermi-Orbital Descriptors". Journal of Computational Chemistry. 40 (32): 2843–2857. doi:10.1002/jcc.26062. ISSN 1096-987X. PMID 31503364. S2CID 202406382.
- Luken, William L.; Culberson, John C. (1984). "Localized orbitals based on the fermi hole". Theoretica Chimica Acta. 66 (5): 279–293. doi:10.1007/BF00554785. eISSN 1432-2234. ISSN 0040-5744. S2CID 96278992.
- Trepte, Kai; Schwalbe, Sebastian; Liebing, Simon; Schulze, Wanja T.; Kortus, Jens; Myneni, Hemanadhan; Ivanov, Aleksei V.; Lehtola, Susi (2021-12-14). "Chemical bonding theories as guides for self-interaction corrected solutions: Multiple local minima and symmetry breaking". The Journal of Chemical Physics. 155 (22): 224109. arXiv:2109.08199. Bibcode:2021JChPh.155v4109T. doi:10.1063/5.0071796. ISSN 0021-9606. PMID 34911315. S2CID 237562804.