Kufor–Rakeb syndrome
Kufor–Rakeb syndrome (KRS) is an autosomal recessive disorder of juvenile onset also known as Parkinson disease-9 (PARK9).[1] It is named after Kufr Rakeb in Irbid, Jordan.[2] Kufor–Rakeb syndrome was first identified in this region in Jordan with a Jordanian couple's 5 children who had rigidity, mask-like face, and bradykinesia.[3] The disease was first described in 1994 by Najim Al-Din et al.[3] The OMIM number is 606693.[3]
| Kufor–Rakeb syndrome | |
|---|---|
| Other names | KRS; Parkinson disease-9 (PARK9) |
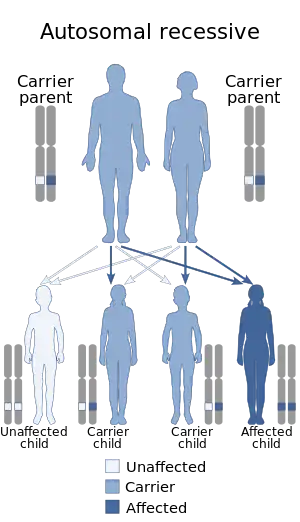 | |
| Autosomal recessive is the manner in which this condition is inherited | |
| Causes | Mutations in the ATP13A2 gene |
| Frequency | <50 individuals have been reported |
Less than 50 individuals have been reported to have KRS.[4] Typically, rapid onset of symptoms occurs between the ages of 12 and 16.[3] It is important to conduct genetic testing to screen family members, so the disease can be detected early and symptoms can be managed.[4]
ATP13A2 gene mutations are associated with Kufor–Rakeb syndrome, first identified in 2010.[2] This syndrome is identified to have a compound heterozygous or homozygous mutation in the ATP13A2 gene.[3] This mutation is located on chromosome 1 and codes for a lysosomal type 5 ATPase.[3] Patients have been identified to have iron in their basal ganglia evident on MRI scans, which has led it to be included as a type of neurodegeneration with brain iron accumulation disorder.[5]
There are no current disease-modifying treatments so treatment focusses on improving symptoms and supportive therapies.[5][6]
Signs and Symptoms
For most individuals with Kufor–Rakeb syndrome, symptoms begin to appear within the first 10 to 20 years of age. Kufor–Rakeb syndrome is a neurodegenerative disorder, so the severity of symptoms tend to progress with time.[6] Symptoms of Kufor–Rakeb syndrome can be divided into two main categories: Motor symptoms (symptoms that affect movement) and non-motor symptoms (symptoms that do not affect movement).
Motor symptoms include:
- Juvenile-onset atypical Parkinsonism (PARK9)[3]
- Bradykinesia (slowed movements)
- Tremors in the chin, tongue, and in some cases the arms
- Rigidity
- Postural instability
- Supranuclear gaze palsy (inability to move eye in a vertical direction)[7]
- Paraplegia (total or partial paralysis of the legs)[6]
- Ataxia (loss of coordination of movements)[6]
- Dystonia (involuntary muscle contractions leading to abnormal postures)[6]
- Dyskinesia (involuntary movements) and hyperreflexia (increased reflexes)
- Facial-faucial-finger mini-myoclonus (involuntary muscle contractions of the fingers, face, and passage at the back of the mouth leading to the pharynx) Non-Motor Symptoms Include:[6]
- Cognitive mental disability and learning difficulty[2]
- Dementia
- Visual and auditory hallucinations
- Severe anxiety and panic attacks
Genetics
Kufor–Rakeb syndrome is associated with mutations in the ATP13A2 gene.[8] The inheritance pattern for KRS is autosomal recessive.[9] If a male and female carrier, who each have one mutation in ATP13A2 have a child, there is a 25% chance the child has KRS, a 50% chance the child is a carrier for KRS, and a 25% chance the child does not have KRS.[7]
The ATP13A2 gene is located on chromosome 1 (1p36.13).[10] The ATP13A2 gene is located in position 36 on the p-arm, which is the short arm, sub-band 13.[11]
The ATP13A2 gene mutations associated with KRS are truncated forms and cause protein instability with loss-of-function.[9] The truncated mutation causes mislocalization of ATP13A2 to the endoplasmic reticulum where the proteasome degrades it through the ER-associated degradation (ERAD) pathway.[9] Heterozygous and homozygous missense mutations in ATP13A2 are linked to early-onset parkinsonism.[9] Compound heterozygous and homozygous mutations in KRS subjects in families from China, Jordan, Pakistan Chile, and Afghanistan, which cause splicing variants or frameshift mutations that truncate ATP13A2.[9] Mutations in ATP13A2 have also been associated with hereditary spastic paraplegia, uncomplicated early- or late-onset parkinsonism, and neuronal ceroid lipofuscinosis.[12]
Diagnosis
Diagnosing Kufor-Rakeb Syndrome requires extensive patient history alongside a physical and neurological examination. KRS can be suspected in individuals with juvenile-onset Parkinsonism in their first 10–20 years of age. In patients with KRS, MRI imaging can show cerebral atrophy and an accumulation of iron in the basal ganglia. This accumulation of iron is referred to as NBIA, or neurodegeneration with brain iron accumulation, and it is a symptom unique to a complex group of inherited neurodegenerative diseases such as KRS, characterized by this accumulation of iron in the basal ganglia.[13] Confirmation is typically through genetic screening for mutations in the ATP13A2 gene.[6]
Management
Currently there are no disease-modifying treatments for Kufor Rakeb syndrome, so therapy is focused on the management of symptoms and improvement of quality of life of affected individuals.[5] Treatment of Kufor Rakeb syndrome is similar to treatment of typical Parkinson's disease and is mainly composed of a combination of two medications called levodopa (L-DOPA) and carbidopa.[14] The goal of this medication is to alleviate motor symptoms by increasing the concentration of dopamine in the nervous system. Dopamine receptor agonists can also be used. Trihexylphenidyl and amantadine might also be prescribed, especially in cases where dopaminergic medication is not effective or tolerated. Botulinum toxin (Botox) can be used to treat dystonia.[6] Medication is only used to control the symptoms of the disease (symptomatic treatment), not to cure it. Benefits of medication are mostly for motor symptoms, and have no significant effect on non-motor symptoms of KRS.[6]
Physical, occupational and/or speech therapy can also be useful interventions. Treatment options for non-motor symptoms are more limited.[15] Individuals living with Kufor Rakeb syndrome might also require a walking aid or a wheelchair. Special education might be indicated, as intellectual disability and learning difficulties are common in KRS. The help of caregivers or health professionals might also be necessary to perform activities of daily living, depending on the severity of the disease. Genetic counselling services should be offered to affected families.
Epidemiology
Kufor–Rakeb syndrome is considered an ultra-rare disorder and has only been diagnosed in less than 50 individuals in literature.[4] KRS is rare, so it is likely that it is often underdiagnosed and the prevalence is higher than is reported in the literature.[4] KRS mutations have been identified in families from China, Jordan, Pakistan Chile, and Afghanistan.[9]
References
- Williams DR, Hadeed A, al-Din AS, Wreikat AL, Lees AJ (October 2005). "Kufor Rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia". Movement Disorders. 20 (10): 1264–71. doi:10.1002/mds.20511. PMID 15986421. S2CID 43558413.
- "Kufor-Rakeb - NBIA Disorders Association". www.nbiadisorders.org. Retrieved 2021-04-26.
- "OMIM Entry - # 606693 - KUFOR-RAKEB SYNDROME; KRS". www.omim.org. Retrieved 2021-04-26.
- "Kufor Rakeb Syndrome". NORD (National Organization for Rare Disorders). Retrieved 2021-05-03.
- Spaull, RVV; Soo, AKS; Hogarth, P; Hayflick, SJ; Kurian, MA (24 November 2021). "Towards Precision Therapies for Inherited Disorders of Neurodegeneration with Brain Iron Accumulation". Tremor and Other Hyperkinetic Movements. 11 (1): 51. doi:10.5334/tohm.661. PMC 8641530. PMID 34909266.accessed 21 April 2022
- "Kufor Rakeb Syndrome". NORD (National Organization for Rare Disorders). Retrieved 2021-04-29.
- "Kufor-Rakeb | NBIA". Retrieved 2021-04-26.
- "Kufor-Rakeb syndrome - MeSH - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2021-05-03.
- Podhajska A, Musso A, Trancikova A, Stafa K, Moser R, Sonnay S, et al. (2012-06-29). "Common pathogenic effects of missense mutations in the P-type ATPase ATP13A2 (PARK9) associated with early-onset parkinsonism". PLOS ONE. 7 (6): e39942. Bibcode:2012PLoSO...739942P. doi:10.1371/journal.pone.0039942. PMC 3386943. PMID 22768177.
- "OMIM Entry * 610513 - ATPase 13A2; ATP13A2". omim.org. Retrieved 2021-05-03.
- "How do geneticists indicate the location of a gene?: MedlinePlus Genetics". medlineplus.gov. Retrieved 2021-05-03.
- Kara E, Tucci A, Manzoni C, Lynch DS, Elpidorou M, Bettencourt C, et al. (July 2016). "Genetic and phenotypic characterization of complex hereditary spastic paraplegia". Brain. 139 (Pt 7): 1904–18. doi:10.1093/brain/aww111. PMC 4939695. PMID 27217339.
- Salomão, Rubens Paulo Araújo; Pedroso, José Luiz; Gama, Maria Thereza Drumond; Dutra, Lívia Almeida; Maciel, Ricardo Horta; Godeiro-Junior, Clécio; Chien, Hsin Fen; Teive, Hélio A. G.; Cardoso, Francisco; Barsottini, Orlando G. P. (July 2016). "A diagnostic approach for neurodegeneration with brain iron accumulation: clinical features, genetics and brain imaging". Arquivos de Neuro-Psiquiatria. 74 (7): 587–596. doi:10.1590/0004-282X20160080. ISSN 1678-4227. PMID 27487380.
- "Levodopa and Carbidopa: MedlinePlus Drug Information". medlineplus.gov. Retrieved 2021-05-02.
- "Physical Therapy for Parkinson's Disease". www.hopkinsmedicine.org. Retrieved 2021-05-02.