Biological small-angle scattering
Biological small-angle scattering is a small-angle scattering method for structure analysis of biological materials. Small-angle scattering is used to study the structure of a variety of objects such as solutions of biological macromolecules, nanocomposites, alloys, and synthetic polymers.[1] Small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS) are the two complementary techniques known jointly as small-angle scattering (SAS).[2] SAS is an analogous method to X-ray and neutron diffraction, wide angle X-ray scattering, as well as to static light scattering. In contrast to other X-ray and neutron scattering methods, SAS yields information on the sizes and shapes of both crystalline and non-crystalline particles. When used to study biological materials, which are very often in aqueous solution, the scattering pattern is orientation averaged.[3][4]
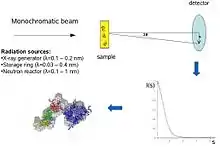
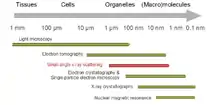
SAS patterns are collected at small angles of a few degrees. SAS is capable of delivering structural information in the resolution range between 1 and 25 nm, and of repeat distances in partially ordered systems of up to 150 nm in size. Ultra small-angle scattering (USAS) can resolve even larger dimensions. The grazing-incidence small-angle scattering (GISAS) is a powerful technique for studying of biological molecule layers on surfaces.
In biological applications SAS is used to determine the structure of a particle in terms of average particle size and shape. One can also get information on the surface-to-volume ratio. Typically, the biological macromolecules are dispersed in a liquid. The method is accurate, mostly non-destructive and usually requires only a minimum of sample preparation. However, biological molecules are always susceptible to radiation damage.
In comparison to other structure determination methods, such as solution NMR or X-ray crystallography, SAS allows one to overcome some restraints. For example, solution NMR is limited to protein size, whereas SAS can be used for small molecules as well as for large multi-molecular assemblies. Solid-State NMR is still an indispensable tool for determining atomic level information of macromolecules greater than 40 kDa or non-crystalline samples such as amyloid fibrils. Structure determination by X-ray crystallography may take several weeks or even years, whereas SAS measurements take days. SAS can also be coupled to other analytical techniques like size-exclusion chromatography to study heterogeneous samples.[5] However, with SAS it is not possible to measure the positions of the atoms within the molecule.
Method
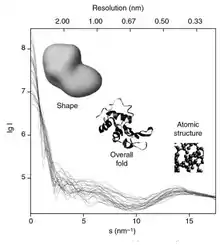
Conceptually, small-angle scattering experiments are simple: the sample is exposed to X-rays or neutrons and the scattered radiation is registered by a detector. As the SAS measurements are performed very close to the primary beam ("small angles"), the technique needs a highly collimated or focused X-ray or neutron beam. The biological small-angle X-ray scattering is often performed at synchrotron radiation sources, because biological molecules normally scatter weakly and the measured solutions are dilute. The biological SAXS method profits from the high intensity of X-ray photon beams provided by the synchrotron storage rings. The X-ray or neutron scattering curve (intensity versus scattering angle) is used to create a low-resolution model of a protein, shown here on the right picture. One can further use the X-ray or neutron scattering data and fit separate domains (X-ray or NMR structures) into the "SAXS envelope".
In a scattering experiment, a solution of macromolecules is exposed to X-rays (with wavelength λ typically around 0.15 nm) or thermal neutrons (λ≈0.5 nm). The scattered intensity I(s) is recorded as a function of momentum transfer s (s=4πsinθ/λ, where 2θ is the angle between the incident and scattered radiation). From the intensity of the solution the scattering from only the solvent is subtracted. The random positions and orientations of particles result in an isotropic intensity distribution which, for monodisperse non-interacting particles, is proportional to the scattering from a single particle averaged over all orientations. The net particle scattering is proportional to the squared difference in scattering length density (electron density for X-rays and nuclear/spin density for neutrons) between particle and solvent – the so-called contrast. The contrast can be varied in neutron scattering using H2O/D2O mixtures or selective deuteration to yield additional information.[1] The information content of SAS data is illustrated here in the figure on the right, which shows X-ray scattering patterns from proteins with different folds and molecular masses. At low angles (2-3 nm resolution) the curves are rapidly decaying functions of s essentially determined by the particle shape, which clearly differ. At medium resolution (2 to 0.5 nm) the differences are already less pronounced and above 0.5 nm resolution all curves are very similar.[6] SAS thus contains information about the gross structural features – shape, quaternary and tertiary structure – but is not suitable for the analysis of the atomic structure.
History
First applications date back to the late 1930s when the main principles of SAXS were developed in the fundamental work of Guinier following his studies of metallic alloys. In the first monograph on SAXS by Guinier and Fournet it was already demonstrated that the method yields not only information on the sizes and shapes of particles but also on the internal structure of disordered and partially ordered systems.
In the 1960s, the method became increasingly important in the study of biological macromolecules in solution as it allowed one to get low-resolution structural information on the overall shape and internal structure in the absence of crystals. A breakthrough in SAXS and SANS experiments came in the 1970s, thanks to the availability of synchrotron radiation and neutron sources, the latter paving the way for contrast variation by solvent exchange of H2O for D2O and specific deuteration methods. It was realised that scattering studies on solution provide, at a minimal investment of time and effort, useful insights into the structure of non-crystalline biochemical systems. Moreover, SAXS/SANS also made possible real time investigations of intermolecular interactions, including assembly and large-scale conformational changes in macromolecular assemblies.
The main difficulty of SAS as a structural method is to extract the three-dimensional structural information of the object from the one-dimensional experimental data. In the past, only overall particle parameters (e.g. volume, radius of gyration) of the macromolecules were directly determined from the experimental data, whereas the analysis in terms of three-dimensional models was limited to simple geometrical bodies (e.g. ellipsoids, cylinders, etc.) or was performed on an ad hoc trial-and-error basis. Electron microscopy was often used as a constraint in building consensus models. In the 1980s, progress in other structural methods led to a decline of the interest of biochemists in SAS studies, which drew structural conclusions from just a couple of overall parameters or were based on trial-and-error models.
The 1990s brought a breakthrough in SAXS/SANS data analysis methods, which opened the way for reliable ab initio modelling of macromolecular complexes, including detailed determination of shape and domain structure and application of rigid body refinement techniques. This progress was accompanied by further advances in instrumentation, allowing sub-ms time resolutions to be achieved on third generation SR sources in the studies of protein and nucleic acid folding.[1]
In 2005, a four-year project was started. Small-Angle X-Ray scattering Initiative for EuRope (SAXIER) with the goal to combine SAXS methods with other analytical techniques and create automated software to rapidly analyse large quantities of data. The project created a unified European SAXS infrastructure, using the most advanced methods available.[7]
Data analysis
In a good quality SAS experiment, several solutions with different concentrations of the macromolecule under investigation are measured. By extrapolating the scattering curves measured at varying concentrations to zero concentration, one is able to get a scattering curve that represents infinite dilution. Then concentration effects should not affect the scattering curve. Data analysis of the extrapolated scattering curve begins with the inspection of the start of the scattering curve in the region around s = 0. If the region follows the Guinier approximation (also known as Guinier law), the sample is not aggregated. Then the shape of the particle in question can be determined by various methods, of which some are described in the following reference.[1]
Indirect Fourier transform
First step is usually to compute a Fourier transform of the scattering curve. Transformed curve can be interpreted as distance distribution function inside a particle. This transformation gives also a benefit of regularization of input data.
Low-resolution models
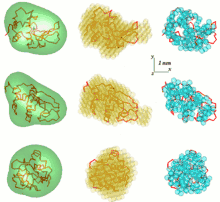
One problem in SAS data analysis is to get a three-dimensional structure from a one-dimensional scattering pattern. The SAS data does not imply a single solution. Many different proteins, for example, may have the same scattering curve. Reconstruction of 3D structure might result in large number of different models. To avoid this problem a number of simplifications need to be considered.
An additional approach is to combine small-angle X-ray and neutron scattering data and model with the program MONSA.
Freely available SAS analysis computer programs have been intensively developed at EMBL. In the first general ab initio approach, an angular envelope function of the particle r=F(ω), where (r,ω) are spherical coordinates, is described by a series of spherical harmonics. The low resolution shape is thus defined by a few parameters – the coefficients of this series – which fit the scattering data. The approach was further developed and implemented in the computer program SASHA (Small Angle Scattering Shape Determination).[8][9] It was demonstrated that under certain circumstances a unique envelope can be extracted from the scattering data. This method is only applicable to globular particles with relatively simple shapes and without significant internal cavities. To overcome these limitations, there was another approach developed, which uses different types of Monte-Carlo searches. DALAI_GA is an elegant program, which takes a sphere with diameter equal to the maximum particle size Dmax, which is determined from the scattering data, and fills it with beads. Each bead belongs either to the particle (index=1) or to the solvent (index=0). The shape is thus described by the binary string of length M. Starting from a random string, a genetic algorithm searches for a model that fits the data. Compactness and connectivity constrains are imposed in the search, implemented in the program DAMMIN.[10][11] If the particle symmetry is known, SASHA and DAMMIN can utilise it as useful constraints. The 'give-n-take' procedure SAXS3D and the program SASMODEL, based on interconnected ellipsoids are ab initio Monte Carlo approaches without limitation in the search space.[6]
An approach that uses an ensemble of Dummy Residues (DRs) and simulated annealing to build a locally "chain-compatible" DR-model inside a sphere of diameter Dmax lets one extract more details from SAXS data. This method is implemented in the program GASBOR.[12][13]
Solution scattering patterns of multi-domain proteins and macromolecular complexes can also be fitted using models built from high resolution (NMR or X-ray) structures of individual domains or subunits assuming that their tertiary structure is preserved. Depending on the complexity of the object, different approaches are employed for the global search of the optimum configuration of subunits fitting the experimental data.
Consensus model
The Monte-Carlo based models contain hundreds or thousand parameters, and caution is required to avoid overinterpretation. A common approach is to align a set of models resulting from independent shape reconstruction runs to obtain an average model retaining the most persistent- and conceivably also most reliable-features (e.g. using the program SUPCOMB).[6][14][15]
Adding missing loops
Disordered surface amino acids ("loops") are frequently unobserved in NMR and crystallographic studies, and may be left missing in the reported models. Such disordered element contribute to the scattering intensity and their probable locations can be found by fixing the known part of the structure and adding the missing parts to fit the SAS pattern from the entire particle. The Dummy Residue approach was extended and the algorithms for adding missing loops or domains were implemented in the program suite CREDO.[6]
Hybrid methods
Recently a few methods proposed that use SAXS data as constraints. The authors aimed to improve results of fold recognition[16] and de novo protein structure prediction[17] methods. SAXS data provide the Fourier transform of the histogram of atomic pair distances (pair distribution function) for a given protein. This can serve as a structural constraint on methods used to determine the native conformational fold of the protein. Threading or fold recognition assumes that 3D structure is more conserved than sequence. Thus, very divergent sequences may have similar structure. Ab initio methods, on the other hand, challenge one of the biggest problems in molecular biology, namely, to predict the folding of a protein "from scratch", using no homologous sequences or structures. Using the "SAXS filter", the authors were able to purify the set of de novo protein models significantly.[17] This was further proved by structure homology searches. It was also shown, that the combination of SAXS scores with scores, used in threading methods, significantly improves the performance of fold recognition.[16] On one example it was demonstrated how approximate tertiary structure of modular proteins can be assembled from high resolution NMR structures of domains, using SAXS data, confining the translational degrees of freedom.[18] Another example shows how the SAXS data can be combined together with NMR, X-ray crystallography and electron microscopy to reconstruct the quaternary structure of multidomain protein.[19]
Flexible systems
An elegant method to tackle the problem of intrinsically disordered or multi-domain proteins with flexible linkers was proposed recently.[20] It allows coexistence of different conformations of a protein, which together contribute to the average experimental scattering pattern. Initially, EOM (ensemble optimization method) generates a pool of models covering the protein configuration space. The scattering curve is then calculated for each model. In the second step, the program selects subsets of protein models. Average experimental scattering is calculated for each subset and fitted to the experimental SAXS data. If the best fit is not found, models are reshuffled between different subsets and new average scattering calculation and fitting to the experimental data is performed. This method has been tested on two proteins– denatured lysozyme and Bruton's protein kinase. It gave some interesting and promising results.[20]
Biological molecule layers and GISAS
Coatings of biomolecules can be studied with grazing-incidence X-ray and neutron scattering. IsGISAXS (grazing incidence small angle X-ray scattering) is a software program dedicated to the simulation and analysis of GISAXS from nanostructures. IsGISAXS only encompasses the scattering by nanometric sized particles, which are buried in a matrix subsurface or supported on a substrate or buried in a thin layer on a substrate. The case of holes is also handled. The geometry is restricted to a plane of particles. The scattering cross section is decomposed in terms of interference function and particle form factor. The emphasis is put on the grazing incidence geometry which induces a "beam refraction effect". The particle form factor is calculated within the distorted wave Born approximation (DWBA), starting as an unperturbed state with sharp interfaces or with the actual perpendicular profile of refraction index. Various kinds of simple geometrical shapes are available with a full account of size and shape distributions in the Decoupling Approximation (DA), in the local monodisperse approximation (LMA) and also in the size-spacing correlation approximation (SSCA). Both, disordered systems of particles defined by their particle-particle pair correlation function and bi-dimensional crystal or para-crystal are considered.[21]
See also
References
- Svergun DI, Koch MH (2003). "Small-angle scattering studies of biological macromolecules in solution". Rep. Prog. Phys. 66 (10): 1735–82. Bibcode:2003RPPh...66.1735S. doi:10.1088/0034-4885/66/10/R05. S2CID 9305500.
- Hamley, I.W. "Small-Angle Scattering: Theory, Instrumentation, Data, and Applications" – Wiley, 2022.
- Ho DL, Byrnes WM, Ma WP, Shi Y, Callaway DJ, Bu Z (September 2004). "Structure-specific DNA-induced conformational changes in Taq polymerase revealed by small angle neutron scattering". The Journal of Biological Chemistry. 279 (37): 39146–54. doi:10.1074/jbc.M404565200. PMID 15247286.
- Lipfert J, Doniach S (1 June 2007). "Small-angle X-ray scattering from RNA, proteins, and protein complexes". Annual Review of Biophysics and Biomolecular Structure. 36 (1): 307–27. doi:10.1146/annurev.biophys.36.040306.132655. PMID 17284163.
- Meisburger SP, Thomas WC, Watkins MB, Ando N (June 2017). "X-ray Scattering Studies of Protein Structural Dynamics". Chemical Reviews. 117 (12): 7615–7672. doi:10.1021/acs.chemrev.6b00790. PMC 5562295. PMID 28558231.
- Svergun DI, Koch MH (October 2002). "Advances in structure analysis using small-angle scattering in solution". Current Opinion in Structural Biology. 12 (5): 654–60. doi:10.1016/S0959-440X(02)00363-9. PMID 12464319.
- "SAXIER: Small-Angle X-ray Scattering Initiative for Europe".
- "SASHA: Small Angle Scattering Shape Determination". Biological small angle scattering group. EMBL Hamburg.
- Svergun DI, Volkov VV, Kozin MB, Stuhrmann HB (1996). "New developments in direct shape determination from small-angle scattering. 2. Uniqueness". Acta Crystallogr. A52 (6): 419–426. doi:10.1107/S0108767391006414.
- "DAMMIN: Ab initio shape determination by simulated annealing using a single phase dummy atom model". Biological small angle scattering group. EMBL Hamburg.
- Svergun DI (June 1999). "Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing". Biophysical Journal. 76 (6): 2879–86. Bibcode:1999BpJ....76.2879S. doi:10.1016/S0006-3495(99)77443-6. PMC 1300260. PMID 10354416.
- "GASBOR: Ab initio reconstruction of a protein structure by a chain-like ensemble of dummy residues". Biological small angle scattering group. EMBL Hamburg.
- Svergun DI, Petoukhov MV, Koch MH (June 2001). "Determination of domain structure of proteins from X-ray solution scattering". Biophysical Journal. 80 (6): 2946–53. Bibcode:2001BpJ....80.2946S. doi:10.1016/S0006-3495(01)76260-1. PMC 1301478. PMID 11371467.
- "SUPCOMB". Biological small angle scattering group. EMBL Hamburg.
- Kozin MB, Svergun DI (2001). "Automated matching of high- and low-resolution structural models". J. Appl. Crystallogr. 34: 33–41. doi:10.1107/S0021889800014126.
- Zheng W, Doniach S (May 2005). "Fold recognition aided by constraints from small angle X-ray scattering data". Protein Engineering, Design & Selection. 18 (5): 209–19. doi:10.1093/protein/gzi026. PMID 15845555.
- Zheng W, Doniach S (February 2002). "Protein structure prediction constrained by solution X-ray scattering data and structural homology identification". Journal of Molecular Biology. 316 (1): 173–87. doi:10.1006/jmbi.2001.5324. PMID 11829511. S2CID 2970219.
- Mattinen ML, Pääkkönen K, Ikonen T, Craven J, Drakenberg T, Serimaa R, Waltho J, Annila A (August 2002). "Quaternary structure built from subunits combining NMR and small-angle x-ray scattering data". Biophysical Journal. 83 (2): 1177–83. Bibcode:2002BpJ....83.1177M. doi:10.1016/S0006-3495(02)75241-7. PMC 1302219. PMID 12124297.
- Tidow H, Melero R, Mylonas E, Freund SM, Grossmann JG, Carazo JM, Svergun DI, Valle M, Fersht AR (July 2007). "Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex". Proceedings of the National Academy of Sciences of the United States of America. 104 (30): 12324–9. Bibcode:2007PNAS..10412324T. doi:10.1073/pnas.0705069104. PMC 1941468. PMID 17620598.
- Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI (May 2007). "Structural characterization of flexible proteins using small-angle X-ray scattering". Journal of the American Chemical Society. 129 (17): 5656–64. doi:10.1021/ja069124n. PMID 17411046.
- "IsGISAXS: a program for analyzing Grazing Incidence Small Angle X-ray Scattering from nanostructures". Archived from the original on May 22, 2012.
Further reading
- Koch MH, Vachette P, Svergun DI (May 2003). "Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution". Quarterly Reviews of Biophysics. 36 (2): 147–227. doi:10.1017/S0033583503003871. PMID 14686102. S2CID 5432509.
- Petoukhov MV, Svergun DI (August 2005). "Global rigid body modeling of macromolecular complexes against small-angle scattering data". Biophysical Journal. 89 (2): 1237–50. Bibcode:2005BpJ....89.1237P. doi:10.1529/biophysj.105.064154. PMC 1366608. PMID 15923225.
- Bernadó P, Blackledge M (December 2010). "Structural biology: Proteins in dynamic equilibrium". Nature. 468 (7327): 1046–8. Bibcode:2010Natur.468.1046B. doi:10.1038/4681046a. PMID 21179158.
External links
- SAXS/WAXS Beamline Australian Synchrotron, Melbourne, Australia
- SIBYLS – beamline at Advanced Light Source, Berkeley, USA
- SAXS – beamline at ELETTRA Synchrotron Light Laboratory, Trieste, Italy
- X33 – beamline at DESY, Hamburg, Germany
- D11A – beamline at Brazilian Synchrotron Light Laboratory, Campinas, Brazil
- X21 and X9 – beamlines at National Synchrotron Light Source at Brookhaven National Laboratory, Upton, USA
- F2 and G1 – beamlines at Cornell Laboratory for Accelerator-based Sciences and Education, Ithaca, USA
- Bio-SANS – beamline at High Flux Isotope Reactor at Oak Ridge National Laboratory, Oak Ridge, TN, USA