Xp11.2 duplication
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23.[1] Most affected females show preferential activation of the duplicated X chromosome.[2] Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication.[3][4] The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).
Symptoms and signs
Information on the clinical symptoms and features is taken from the Human Phenotype Ontology database[5] and the Unique database.[1] All affected individuals needn't show all the symptoms. Some of the most noted features are:
- Intellectual and Learning Disabilities
- Speech delay
- Early puberty
- Significant weight and height problems
- Lower-extremity anomalies (anomalies of the legs and/ or feet)
- Epileptic Seizures
- Unusual pattern of EEG with centro-temporal focal spike waves in children
- Minor facial features
Intellectual and Learning Disabilities
Among people who need support with their learning, at least 3% are believed to carry the duplication.[3] It is noted that affected members of the same family with the same Xp11.2 microduplication generally have similar learning profiles. Children with small duplications of 0.5-1.3 Mb seem to have a mild learning difficulty, while others with the typical duplication of around 4.5 Mb generally have a borderline, mild or moderate learning disability. An extreme case with a very large duplication of 55 Mb has shown to have a severe intellectual disability.[3][6]
Speech Delay
Speech is very commonly affected and usually the first sign. Both speech and comprehension seem to be affected, to various degrees of extent. Low facial muscle tone underlies difficulty in making certain sounds of speech. Nasal or hoarse voice are also observed. Babies are known to be unable to suck from breast in their infancy due to weak facial muscles.
Early Puberty
Early puberty occurs in 80% of the affected children or adults, with girls starting their menstrual cycles as early as age 9 and boys showing signs of puberty at age 8.5. One boy from Unique had completed puberty by age 13.[1]
Weight problems
Affected children show tendency to be overweight. This might indicate metabolism issues.
Lower-extremity Anomalies
Anomalies of lower limbs or feet are common in people with an Xp11.2 duplication, affecting about 71% cases. Features include flat feet, arched feet (pes cavus), clubfoot (talipes), narrow feet, webbed or joined toes/fingers (syndactyly), 5th finger clinodactyly, 5th toe hypoplasia, and tapering fingers.[3][7]
Unusual EEG Pattern
A typical pattern of electrical activity in the brain of affected children, described as ‘subclinical seizures’ has been noted. A peculiar electroencephalographic pattern characterized by rolandic-like spikes and/or continuous spike wave during slow sleep (CSWS), also called centro-temporal focal spike, exists in childhood.[3]
Cause
Using array-based Comparative Genomic Hybridization (aCGH) to screen 2,400 individuals with isolated or syndromic mental retardation for copy number variation, Giorda et al. (2009) identified 8 (0.33%) unrelated individuals, 2 males and 6 females, with a microduplication at chromosome Xp11.23-p11.22. The rearrangement was familial in 3 patients. A female patient shared a 4.5-Mb duplication with her affected mother and sister, and an unrelated male patient shared a 4.5-Mb duplication with his affected mother and sister. A third unrelated male inherited a smaller 0.8-Mb duplication from his unaffected mother. Three additional individuals had de novo 4.5-Mb duplications, and 2 more had partially overlapping de novo 6.0- and 9.2-Mb duplications. Paternal origin of the duplication was demonstrated in all de novo female cases. Six affected females had selective inactivation of the normal X chromosome, whereas 3 had random X inactivation. Breakpoints could be identified in 8 individuals. The recurrent duplication was flanked distally by a segmental duplication (D-REP at 47.8-48.2 Mb) containing a cluster of genes and pseudogenes of the synovial sarcoma X breakpoint (SSX) and proximally by a complex repeat (P-REP at 52.1-53.1 Mb) rich in SSX, melanoma antigen and X antigen (XAGE) genes. Sequence analysis of the junctions demonstrated that the recurrent 4.5-Mb duplications were mediated by non-allelic homologous recombination (NAHR) or Alu-mediated recombination. The majority of these recombinations occurred between flanking complex segmental duplications.[3] Region of duplication and copy number variation can be further confirmed by Fluorescence In-Situ Hybridization (FISH) and PCR.
Genetics
The duplication at Xp11.2, especially the Xp11.22-11.23 region is syndromic and is implicated in X-linked mental retardation.[8][4] The chromosomal duplication can be de novo or familial. Familial carriers of small duplication (<1 Mb) show X-linked recessive inheritance. All other affected individuals with larger duplication present dominant expression and comparable clinical phenotypes irrespective of sex, duplication size, and X-inactivation pattern.[3]
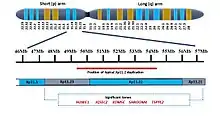
Xp11.22 comprises approximately 5 Mb of DNA (chrX:49,800,001–54,800,000, hg19). A number of pathogenic deletions and duplications involving Xp11.22 have been described in individuals with developmental delay, intellectual disability and/or autism.[9] These phenotypes have been attributed to changes in the copy number of several genes including HUWE1, KDM5C, IQSEC2, TSPYL2, SHROOM4, PHF8 and FAM120C.[10][11]
HUWE1
The HECT, UBA and WWE domain-containing protein 1 (HUWE1) is a HECT family ubiquitin ligase located on the X chromosome at Xp11.22 with growing genetic links to cancer[12] and intellectual disability.[13] Expression of the HUWE1 gene is found in several mouse tissues including cortex, hippocampus, tongue, eye, kidney, liver, adrenal gland, and fibroblasts.[14] Increased copies of HUWE1 are associated with non-syndromic intellectual disability.[14][15] Missense mutations in HUWE1 occur in multiple families with intellectual disability, including families with Juberg-Marsidi-Brooks syndrome.[15][13][16] Patients with missense mutations in HUWE1 share clinical features with patients with a duplication of HUWE1. This suggests both increased and decreased HUWE1 function could be associated with intellectual disability, but evidence from an in vivo model system supporting or refuting this possibility remains absent.
KDM5C
KDM5C (Lysine-Specific Demethylase 5C) also known as jumonji, A/T-rich interactive domain 1C (JARID1C) is located on the X chromosome at Xp11.22-p11.21. The gene encodes a 1560 amino-acid protein that belongs to the JARID1 subfamily of Arid DNA-binding proteins.[17] The protein possesses H3K4me3-specific demethylase activity and is shown to function as a transcriptional repressor through the RE-1-silencing transcription factor (REST) complex.[18]
The mutations in KDM5C cause Claes-Jensen type[19][20] syndromic X-linked Intellectual Disability characterized by moderate-to-severe ID, speech abnormalities and other clinical findings such as seizures and aggressive behavior in some individuals.[11][21] There is also a report of a mutation in a patient with autism spectrum disorder.[22] A study showed that Kdm5c-knockout mice exhibit adaptive and cognitive abnormalities similar to those in human X-linked intellectual disability and concluded that histone methylation dynamics sculpt the neuronal network.[23]
IQSEC2
IQ motif and Sec7 domain 2 (IQSEC2), also known as BRAG1 or IQ-ARFGEF, is located on the X chromosome at Xp11.22 and encodes guanine nucleotide exchange factor for the ARF family of GTP-binding proteins (ARFGEF).[24] It is expressed in the neurons and is involved in cytoskeletal organization, dendritic spine morphology, and excitatory synaptic organization.[25]
Mutations in IQSEC2 are widely associated in cases of X-linked non-syndromic mental retardation, with some carrier females reported with learning disabilities.[26] This gene is known to play a significant role in the maintenance of homeostasis within the neural environment of the human brain. A change of guanine nucleotide exchange factor activity may influence the regulation of actin cytoskeleton organization and neuronal development in the brain by reduced activation of the ARF6 substrate or a defect in the GTP-binding activity.[26]
Two intragenic duplications predicted to cause termination mutations on the X-chromosome involving IQSEC2 were identified in two de novo cases, and one nonsense mutation was described in three additional male patients presenting severe intellectual disability and additional clinical features including neonatal hypotonia, delayed motor skills, seizures, strabismus, autistic-like behavior, stereotypic midline hand movements, microcephaly, little-to-no walking, little-to-no language skills, significant behavioral issues, and mildly abnormal facial features.[27] A novel de novo mutation in the IQSEC2 gene identified through diagnostic exome sequencing showed significant developmental delay, seizures, hypotonia, vision impairments, plagiocephaly, autistic-like features, absent language skills, and abnormal MRI findings.[28] IQSEC2 gene plays a larger role in the cause of X-linked cognitive impairment than previously thought. Additional consideration is warranted with regards to the syndromic nature of its phenotypic association.
TSPYL2
Testis-Specific Protein Y-encoded (TSPY) Like 2 (TSPYL2) codes for a member of the TSPY-like/SET/nucleosome assembly protein-1 superfamily and is located on the X chromosome at Xp11.22. The encoded protein is localized to the nucleolus where it functions in chromatin remodeling and as an inhibitor of cell-cycle progression.[29] Consistent with a possible role for Tspyl2 pathways in neurodevelopment, Xp11.2 microduplication incorporating the TSPYL2 locus has been reported in male patients with Attention Deficit Hyperactivity Disorder.[11]
SHROOM4
Shroom Family Member 4 (SHROOM4), also known as KIAA1202, encodes a member of the APX/Shroom family, which contains an N-terminal PDZ domain and a C-terminal ASD2 motif. It is located on the X chromosome at Xp11.22 and is mainly associated with the Stocco dos Santos X-linked mental retardation syndrome characterized by cognitive disabilities.[30] The encoded protein may play a role in cytoskeletal architecture. Symptoms of SHROOM4 gene mutations in the original family described by Stocco dos Santos include severe intellectual disability, bilateral congenital hip luxation and short stature. The SHROOM4 gene was also found to be disrupted in two unrelated females with mild to moderate intellectual disabilities. Other features included delayed or no speech, seizures, kyphosis and hyperactivity. Carrier females displayed seizures and depression. No mutations in SHROOM4 were identified in more than 1000 control X chromosomes.[30][31]
See also
References
- "Unique The Rare Chromosome Disorder Support Group". www.rarechromo.org. Retrieved 2018-02-28.
- "Microduplication Xp11.22-p11.23 syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-02-28.
- Giorda R, Bonaglia MC, Beri S, Fichera M, Novara F, Magini P, et al. (September 2009). "Complex segmental duplications mediate a recurrent dup(X)(p11.22-p11.23) associated with mental retardation, speech delay, and EEG anomalies in males and females". American Journal of Human Genetics. 85 (3): 394–400. doi:10.1016/j.ajhg.2009.08.001. PMC 2771536. PMID 19716111.
- Honda S, Hayashi S, Imoto I, Toyama J, Okazawa H, Nakagawa E, Goto Y, Inazawa J (September 2010). "Copy-number variations on the X chromosome in Japanese patients with mental retardation detected by array-based comparative genomic hybridization analysis". Journal of Human Genetics. 55 (9): 590–9. doi:10.1038/jhg.2010.74. PMID 20613765.
- "Home". Human Phenotype Ontology. Retrieved 2018-02-28.
- Nizon M, Andrieux J, Rooryck C, de Blois MC, Bourel-Ponchel E, Bourgois B, et al. (January 2015). "Phenotype-genotype correlations in 17 new patients with an Xp11.23p11.22 microduplication and review of the literature". American Journal of Medical Genetics. Part A. 167A (1): 111–22. doi:10.1002/ajmg.a.36807. PMID 25425167. S2CID 32958511.
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP (April 2009). "DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources". American Journal of Human Genetics. 84 (4): 524–33. doi:10.1016/j.ajhg.2009.03.010. PMC 2667985. PMID 19344873.
- Online Mendelian Inheritance in Man (OMIM): Chromosome Xp11.22 Duplication Syndrome - 300705
- Grau C, Starkovich M, Azamian MS, Xia F, Cheung SW, Evans P, Henderson A, Lalani SR, Scott DA (2017-04-17). "Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability". PLOS ONE. 12 (4): e0175962. Bibcode:2017PLoSO..1275962G. doi:10.1371/journal.pone.0175962. PMC 5393878. PMID 28414775.
- Orivoli S, Pavlidis E, Cantalupo G, Pezzella M, Zara F, Garavelli L, Pisani F, Piccolo B (January 2016). "Xp11.22 Microduplications Including HUWE1: Case Report and Literature Review". Neuropediatrics. 47 (1): 51–6. doi:10.1055/s-0035-1566233. PMID 26587761. S2CID 38240788.
- Moey C, Hinze SJ, Brueton L, Morton J, McMullan DJ, Kamien B, Barnett CP, Brunetti-Pierri N, Nicholl J, Gecz J, Shoubridge C (March 2016). "Xp11.2 microduplications including IQSEC2, TSPYL2 and KDM5C genes in patients with neurodevelopmental disorders". European Journal of Human Genetics. 24 (3): 373–80. doi:10.1038/ejhg.2015.123. PMC 4757771. PMID 26059843.
- Zhong Q, Gao W, Du F, Wang X (July 2005). "Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis". Cell. 121 (7): 1085–95. doi:10.1016/j.cell.2005.06.009. PMID 15989957.
- Froyen G, Corbett M, Vandewalle J, Jarvela I, Lawrence O, Meldrum C, et al. (February 2008). "Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation". American Journal of Human Genetics. 82 (2): 432–43. doi:10.1016/j.ajhg.2007.11.002. PMC 2426915. PMID 18252223.
- Froyen G, Belet S, Martinez F, Santos-Rebouças CB, Declercq M, Verbeeck J, et al. (August 2012). "Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements". American Journal of Human Genetics. 91 (2): 252–64. doi:10.1016/j.ajhg.2012.06.010. PMC 3415555. PMID 22840365.
- Friez MJ, Brooks SS, Stevenson RE, Field M, Basehore MJ, Adès LC, et al. (April 2016). "HUWE1 mutations in Juberg-Marsidi and Brooks syndromes: the results of an X-chromosome exome sequencing study". BMJ Open. 6 (4): e009537. doi:10.1136/bmjopen-2015-009537. PMC 4854010. PMID 27130160.
- Isrie M, Kalscheuer VM, Holvoet M, Fieremans N, Van Esch H, Devriendt K (July 2013). "HUWE1 mutation explains phenotypic severity in a case of familial idiopathic intellectual disability". European Journal of Medical Genetics. 56 (7): 379–82. doi:10.1016/j.ejmg.2013.05.005. hdl:11858/00-001M-0000-0018-EE0C-9. PMID 23721686.
- Kortschak RD, Tucker PW, Saint R (June 2000). "ARID proteins come in from the desert". Trends in Biochemical Sciences. 25 (6): 294–9. doi:10.1016/S0968-0004(00)01597-8. PMID 10838570.
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y (March 2007). "The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases". Cell. 128 (6): 1077–88. doi:10.1016/j.cell.2007.02.017. PMID 17320160.
- Claes S, Devriendt K, Van Goethem G, Roelen L, Meireleire J, Raeymaekers P, Cassiman JJ, Fryns JP (September 2000). "Novel syndromic form of X-linked complicated spastic paraplegia". American Journal of Medical Genetics. 94 (1): 1–4. doi:10.1002/1096-8628(20000904)94:1<1::AID-AJMG1>3.0.CO;2-V. PMID 10982473.
- Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, et al. (February 2005). "Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation". American Journal of Human Genetics. 76 (2): 227–36. doi:10.1086/427563. PMC 1196368. PMID 15586325.
- Rujirabanjerd S, Nelson J, Tarpey PS, Hackett A, Edkins S, Raymond FL, Schwartz CE, Turner G, Iwase S, Shi Y, Futreal PA, Stratton MR, Gecz J (March 2010). "Identification and characterization of two novel JARID1C mutations: suggestion of an emerging genotype-phenotype correlation". European Journal of Human Genetics. 18 (3): 330–5. doi:10.1038/ejhg.2009.175. PMC 2987212. PMID 19826449.
- Adegbola A, Gao H, Sommer S, Browning M (February 2008). "A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD)". American Journal of Medical Genetics. Part A. 146A (4): 505–11. doi:10.1002/ajmg.a.32142. PMID 18203167. S2CID 31730709.
- Iwase S, Brookes E, Agarwal S, Badeaux AI, Ito H, Vallianatos CN, Tomassy GS, Kasza T, Lin G, Thompson A, Gu L, Kwan KY, Chen C, Sartor MA, Egan B, Xu J, Shi Y (February 2016). "A Mouse Model of X-linked Intellectual Disability Associated with Impaired Removal of Histone Methylation". Cell Reports. 14 (5): 1000–1009. doi:10.1016/j.celrep.2015.12.091. PMC 4749408. PMID 26804915.
- Nagase T, Ishikawa K, Suyama M, Kikuno R, Hirosawa M, Miyajima N, Tanaka A, Kotani H, Nomura N, Ohara O (February 1999). "Prediction of the coding sequences of unidentified human genes. XIII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro". DNA Research. 6 (1): 63–70. doi:10.1093/dnares/6.1.63. PMID 10231032.
- Kalscheuer VM, James VM, Himelright ML, Long P, Oegema R, Jensen C, Bienek M, Hu H, Haas SA, Topf M, Hoogeboom AJ, Harvey K, Walikonis R, Harvey RJ (2015). "Novel Missense Mutation A789V in IQSEC2 Underlies X-Linked Intellectual Disability in the MRX78 Family". Frontiers in Molecular Neuroscience. 8: 85. doi:10.3389/fnmol.2015.00085. PMC 4707274. PMID 26793055.
- Shoubridge C, Tarpey PS, Abidi F, Ramsden SL, Rujirabanjerd S, Murphy JA, et al. (June 2010). "Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability". Nature Genetics. 42 (6): 486–8. doi:10.1038/ng.588. PMC 3632837. PMID 20473311.
- Tran Mau-Them F, Willems M, Albrecht B, Sanchez E, Puechberty J, Endele S, Schneider A, Ruiz Pallares N, Missirian C, Rivier F, Girard M, Holder M, Manouvrier S, Touitou I, Lefort G, Sarda P, Moncla A, Drunat S, Wieczorek D, Genevieve D (February 2014). "Expanding the phenotype of IQSEC2 mutations: truncating mutations in severe intellectual disability". European Journal of Human Genetics. 22 (2): 289–92. doi:10.1038/ejhg.2013.113. PMC 3895633. PMID 23674175.
- Gandomi SK, Farwell Gonzalez KD, Parra M, Shahmirzadi L, Mancuso J, Pichurin P, Temme R, Dugan S, Zeng W, Tang S (June 2014). "Diagnostic exome sequencing identifies two novel IQSEC2 mutations associated with X-linked intellectual disability with seizures: implications for genetic counseling and clinical diagnosis". Journal of Genetic Counseling. 23 (3): 289–98. doi:10.1007/s10897-013-9671-6. PMID 24306141. S2CID 17850707.
- "TSPYL2 TSPY like 2 [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov. Retrieved 2018-03-01.
- dos Santos RC, Barretto OC, Nonoyama K, Castro NH, Ferraz OP, Walter-Moura J, Vescio CC, Beçak W (May 1991). "X-linked syndrome: mental retardation, hip luxation, and G6PD variant [Gd(+) Butantan]". American Journal of Medical Genetics. 39 (2): 133–6. doi:10.1002/ajmg.1320390204. PMID 2063914.
- Stocco dos Santos RC, Castro NH, Lillia Holmes A, Beçak W, Tackels-Horne D, Lindsey CJ, Lubs HA, Stevenson RE, Schwartz CE (April 2003). "Stocco dos Santos X-linked mental retardation syndrome: clinical elucidation and localization to Xp11.3-Xq21.3". American Journal of Medical Genetics. Part A. 118A (3): 255–9. doi:10.1002/ajmg.a.20021. PMID 12673656. S2CID 32244491.