Structural alignment
Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.
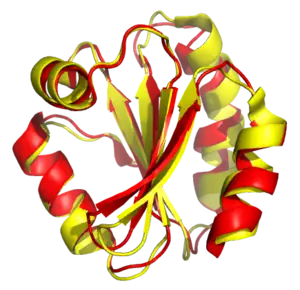
Structural alignments can compare two sequences or multiple sequences. Because these alignments rely on information about all the query sequences' three-dimensional conformations, the method can only be used on sequences where these structures are known. These are usually found by X-ray crystallography or NMR spectroscopy. It is possible to perform a structural alignment on structures produced by structure prediction methods. Indeed, evaluating such predictions often requires a structural alignment between the model and the true known structure to assess the model's quality.[1] Structural alignments are especially useful in analyzing data from structural genomics and proteomics efforts, and they can be used as comparison points to evaluate alignments produced by purely sequence-based bioinformatics methods.[2][3][4]
The outputs of a structural alignment are a superposition of the atomic coordinate sets and a minimal root mean square deviation (RMSD) between the structures. The RMSD of two aligned structures indicates their divergence from one another. Structural alignment can be complicated by the existence of multiple protein domains within one or more of the input structures, because changes in relative orientation of the domains between two structures to be aligned can artificially inflate the RMSD.
Data produced by structural alignment
The minimum information produced from a successful structural alignment is a set of residues that are considered equivalent between the structures. This set of equivalences is then typically used to superpose the three-dimensional coordinates for each input structure. (Note that one input element may be fixed as a reference and therefore its superposed coordinates do not change.) The fitted structures can be used to calculate mutual RMSD values, as well as other more sophisticated measures of structural similarity such as the global distance test (GDT,[5] the metric used in CASP). The structural alignment also implies a corresponding one-dimensional sequence alignment from which a sequence identity, or the percentage of residues that are identical between the input structures, can be calculated as a measure of how closely the two sequences are related.
Types of comparisons
Because protein structures are composed of amino acids whose side chains are linked by a common protein backbone, a number of different possible subsets of the atoms that make up a protein macromolecule can be used in producing a structural alignment and calculating the corresponding RMSD values. When aligning structures with very different sequences, the side chain atoms generally are not taken into account because their identities differ between many aligned residues. For this reason it is common for structural alignment methods to use by default only the backbone atoms included in the peptide bond. For simplicity and efficiency, often only the alpha carbon positions are considered, since the peptide bond has a minimally variant planar conformation. Only when the structures to be aligned are highly similar or even identical is it meaningful to align side-chain atom positions, in which case the RMSD reflects not only the conformation of the protein backbone but also the rotameric states of the side chains. Other comparison criteria that reduce noise and bolster positive matches include secondary structure assignment, native contact maps or residue interaction patterns, measures of side chain packing, and measures of hydrogen bond retention.[6]
Structural superposition
The most basic possible comparison between protein structures makes no attempt to align the input structures and requires a precalculated alignment as input to determine which of the residues in the sequence are intended to be considered in the RMSD calculation. Structural superposition is commonly used to compare multiple conformations of the same protein (in which case no alignment is necessary, since the sequences are the same) and to evaluate the quality of alignments produced using only sequence information between two or more sequences whose structures are known. This method traditionally uses a simple least-squares fitting algorithm, in which the optimal rotations and translations are found by minimizing the sum of the squared distances among all structures in the superposition.[7] More recently, maximum likelihood and Bayesian methods have greatly increased the accuracy of the estimated rotations, translations, and covariance matrices for the superposition.[8][9]
Algorithms based on multidimensional rotations and modified quaternions have been developed to identify topological relationships between protein structures without the need for a predetermined alignment. Such algorithms have successfully identified canonical folds such as the four-helix bundle.[10] The SuperPose method is sufficiently extensible to correct for relative domain rotations and other structural pitfalls.[11]
Evaluating similarity
Often the purpose of seeking a structural superposition is not so much the superposition itself, but an evaluation of the similarity of two structures or a confidence in a remote alignment.[1][2][3] A subtle but important distinction from maximal structural superposition is the conversion of an alignment to a meaningful similarity score.[12][13] Most methods output some sort of "score" indicating the quality of the superposition.[5][14][15][12][13] However, what one actually wants is not merely an estimated "Z-score" or an estimated E-value of seeing the observed superposition by chance but instead one desires that the estimated E-value is tightly correlation to the true E-value. Critically, even if a method's estimated E-value is precisely correct on average, if it lacks a low standard deviation on its estimated value generation process, then the rank ordering of the relative similarities of a query protein to a comparison set will rarely agree with the "true" ordering.[12][13]
Different methods will superimpose different numbers of residues because they use different quality assurances and different definitions of "overlap"; some only include residues meeting multiple local and global superposition criteria and others are more greedy, flexible, and promiscuous. A greater number of atoms superposed can mean more similarity but it may not always produce the best E-value quantifying the unlikeliness of the superposition and thus not as useful for assessing similarity, especially in remote homologs.[1][2][3][4]
Algorithmic complexity
Optimal solution
The optimal "threading" of a protein sequence onto a known structure and the production of an optimal multiple sequence alignment have been shown to be NP-complete.[16][17] However, this does not imply that the structural alignment problem is NP-complete. Strictly speaking, an optimal solution to the protein structure alignment problem is only known for certain protein structure similarity measures, such as the measures used in protein structure prediction experiments, GDT_TS[5] and MaxSub.[14] These measures can be rigorously optimized using an algorithm capable of maximizing the number of atoms in two proteins that can be superimposed under a predefined distance cutoff.[15] Unfortunately, the algorithm for optimal solution is not practical, since its running time depends not only on the lengths but also on the intrinsic geometry of input proteins.
Approximate solution
Approximate polynomial-time algorithms for structural alignment that produce a family of "optimal" solutions within an approximation parameter for a given scoring function have been developed.[15][18] Although these algorithms theoretically classify the approximate protein structure alignment problem as "tractable", they are still computationally too expensive for large-scale protein structure analysis. As a consequence, practical algorithms that converge to the global solutions of the alignment, given a scoring function, do not exist. Most algorithms are, therefore, heuristic, but algorithms that guarantee the convergence to at least local maximizers of the scoring functions, and are practical, have been developed.[19]
Representation of structures
Protein structures have to be represented in some coordinate-independent space to make them comparable. This is typically achieved by constructing a sequence-to-sequence matrix or series of matrices that encompass comparative metrics: rather than absolute distances relative to a fixed coordinate space. An intuitive representation is the distance matrix, which is a two-dimensional matrix containing all pairwise distances between some subset of the atoms in each structure (such as the alpha carbons). The matrix increases in dimensionality as the number of structures to be simultaneously aligned increases. Reducing the protein to a coarse metric such as secondary structure elements (SSEs) or structural fragments can also produce sensible alignments, despite the loss of information from discarding distances, as noise is also discarded.[20] Choosing a representation to facilitate computation is critical to developing an efficient alignment mechanism.
Methods
Structural alignment techniques have been used in comparing individual structures or sets of structures as well as in the production of "all-to-all" comparison databases that measure the divergence between every pair of structures present in the Protein Data Bank (PDB). Such databases are used to classify proteins by their fold.
DALI
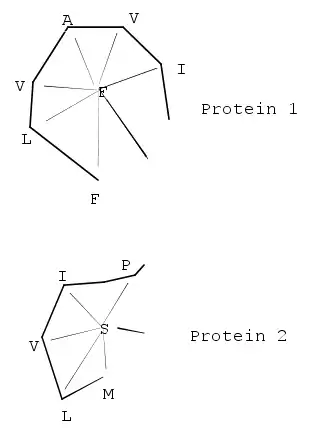
A common and popular structural alignment method is the DALI, or Distance-matrix ALIgnment method, which breaks the input structures into hexapeptide fragments and calculates a distance matrix by evaluating the contact patterns between successive fragments.[21] Secondary structure features that involve residues that are contiguous in sequence appear on the matrix's main diagonal; other diagonals in the matrix reflect spatial contacts between residues that are not near each other in the sequence. When these diagonals are parallel to the main diagonal, the features they represent are parallel; when they are perpendicular, their features are antiparallel. This representation is memory-intensive because the features in the square matrix are symmetrical (and thus redundant) about the main diagonal.
When two proteins' distance matrices share the same or similar features in approximately the same positions, they can be said to have similar folds with similar-length loops connecting their secondary structure elements. DALI's actual alignment process requires a similarity search after the two proteins' distance matrices are built; this is normally conducted via a series of overlapping submatrices of size 6x6. Submatrix matches are then reassembled into a final alignment via a standard score-maximization algorithm — the original version of DALI used a Monte Carlo simulation to maximize a structural similarity score that is a function of the distances between putative corresponding atoms. In particular, more distant atoms within corresponding features are exponentially downweighted to reduce the effects of noise introduced by loop mobility, helix torsions, and other minor structural variations.[20] Because DALI relies on an all-to-all distance matrix, it can account for the possibility that structurally aligned features might appear in different orders within the two sequences being compared.
The DALI method has also been used to construct a database known as FSSP (Fold classification based on Structure-Structure alignment of Proteins, or Families of Structurally Similar Proteins) in which all known protein structures are aligned with each other to determine their structural neighbors and fold classification. There is an searchable database based on DALI as well as a downloadable program and web search based on a standalone version known as DaliLite.
Combinatorial extension
The combinatorial extension (CE) method is similar to DALI in that it too breaks each structure in the query set into a series of fragments that it then attempts to reassemble into a complete alignment. A series of pairwise combinations of fragments called aligned fragment pairs, or AFPs, are used to define a similarity matrix through which an optimal path is generated to identify the final alignment. Only AFPs that meet given criteria for local similarity are included in the matrix as a means of reducing the necessary search space and thereby increasing efficiency.[22] A number of similarity metrics are possible; the original definition of the CE method included only structural superpositions and inter-residue distances but has since been expanded to include local environmental properties such as secondary structure, solvent exposure, hydrogen-bonding patterns, and dihedral angles.[22]
An alignment path is calculated as the optimal path through the similarity matrix by linearly progressing through the sequences and extending the alignment with the next possible high-scoring AFP pair. The initial AFP pair that nucleates the alignment can occur at any point in the sequence matrix. Extensions then proceed with the next AFP that meets given distance criteria restricting the alignment to low gap sizes. The size of each AFP and the maximum gap size are required input parameters but are usually set to empirically determined values of 8 and 30 respectively.[22] Like DALI and SSAP, CE has been used to construct an all-to-all fold classification database Archived 1998-12-03 at the Wayback Machine from the known protein structures in the PDB.
The RCSB PDB has recently released an updated version of CE, Mammoth, and FATCAT as part of the RCSB PDB Protein Comparison Tool. It provides a new variation of CE that can detect circular permutations in protein structures.[23]
Mammoth
MAMMOTH [12] approaches the alignment problem from a different objective than almost all other methods. Rather than trying to find an alignment that maximally superimposes the largest number of residues, it seeks the subset of the structural alignment least likely to occur by chance. To do this it marks a local motif alignment with flags to indicate which residues simultaneously satisfy more stringent criteria: 1) Local structure overlap 2) regular secondary structure 3) 3D-superposition 4) same ordering in primary sequence. It converts the statistics of the number of residues with high-confidence matches and the size of the protein to compute an Expectation value for the outcome by chance. It excels at matching remote homologs, particularly structures generated by ab initio structure prediction to structure families such as SCOP, because it emphasizes extracting a statistically reliable sub alignment and not in achieving the maximal sequence alignment or maximal 3D superposition.[2][3]
For every overlapping window of 7 consecutive residues it computes the set of displacement direction unit vectors between adjacent C-alpha residues. All-against-all local motifs are compared based on the URMS score. These values becomes the pair alignment score entries for dynamic programming which produces a seed pair-wise residue alignment. The second phase uses a modified MaxSub algorithm: a single 7 reside aligned pair in each proteins is used to orient the two full length protein structures to maximally superimpose these just these 7 C-alpha, then in this orientation it scans for any additional aligned pairs that are close in 3D. It re-orients the structures to superimpose this expanded set and iterates until no more pairs coincide in 3D. This process is restarted for every 7 residue window in the seed alignment. The output is the maximal number of atoms found from any of these initial seeds. This statistic is converted to a calibrated E-value for the similarity of the proteins.
Mammoth makes no attempt to re-iterate the initial alignment or extend the high quality sub-subset. Therefore, the seed alignment it displays can't be fairly compared to DALI or TM align as it was formed simply as a heuristic to prune the search space. (It can be used if one wants an alignment based solely on local structure-motif similarity agnostic of long range rigid body atomic alignment.) Because of that same parsimony, it is well over ten times faster than DALI, CE and TM-align.[24] It is often used in conjunction with these slower tools to pre-screen large data bases to extract the just the best E-value related structures for more exhaustive superposition or expensive calculations. [25] [26]
It has been particularly successful at analyzing "decoy" structures from ab initio structure prediction.[1][2][3] These decoys are notorious for getting local fragment motif structure correct, and forming some kernels of correct 3D tertiary structure but getting the full length tertiary structure wrong. In this twilight remote homology regime, Mammoth's e-values for the CASP[1] protein structure prediction evaluation have been shown to be significantly more correlated with human ranking than SSAP or DALI.[12] Mammoths ability to extract the multi-criteria partial overlaps with proteins of known structure and rank these with proper E-values, combined with its speed facilitates scanning vast numbers of decoy models against the PDB data base for identifying the most likely correct decoys based on their remote homology to known proteins. [2]
SSAP
The SSAP (Sequential Structure Alignment Program) method uses double dynamic programming to produce a structural alignment based on atom-to-atom vectors in structure space. Instead of the alpha carbons typically used in structural alignment, SSAP constructs its vectors from the beta carbons for all residues except glycine, a method which thus takes into account the rotameric state of each residue as well as its location along the backbone. SSAP works by first constructing a series of inter-residue distance vectors between each residue and its nearest non-contiguous neighbors on each protein. A series of matrices are then constructed containing the vector differences between neighbors for each pair of residues for which vectors were constructed. Dynamic programming applied to each resulting matrix determines a series of optimal local alignments which are then summed into a "summary" matrix to which dynamic programming is applied again to determine the overall structural alignment.
SSAP originally produced only pairwise alignments but has since been extended to multiple alignments as well.[27] It has been applied in an all-to-all fashion to produce a hierarchical fold classification scheme known as CATH (Class, Architecture, Topology, Homology),[28] which has been used to construct the CATH Protein Structure Classification database.
Recent developments
Improvements in structural alignment methods constitute an active area of research, and new or modified methods are often proposed that are claimed to offer advantages over the older and more widely distributed techniques. A recent example, TM-align, uses a novel method for weighting its distance matrix, to which standard dynamic programming is then applied.[29][13] The weighting is proposed to accelerate the convergence of dynamic programming and correct for effects arising from alignment lengths. In a benchmarking study, TM-align has been reported to improve in both speed and accuracy over DALI and CE.[29]
Other promising methods of structural alignment are local structural alignment methods. These provide comparison of pre-selected parts of proteins (e.g. binding sites, user-defined structural motifs) [30][31][32] against binding sites or whole-protein structural databases. The MultiBind and MAPPIS servers [32][33] allow the identification of common spatial arrangements of physicochemical properties such as H-bond donor, acceptor, aliphatic, aromatic or hydrophobic in a set of user provided protein binding sites defined by interactions with small molecules (MultiBind) or in a set of user-provided protein–protein interfaces (MAPPIS). Others provide comparison of entire protein structures [34] against a number of user submitted structures or against a large database of protein structures in reasonable time (ProBiS[35]). Unlike global alignment approaches, local structural alignment approaches are suited to detection of locally conserved patterns of functional groups, which often appear in binding sites and have significant involvement in ligand binding.[33] As an example, comparing G-Losa,[36] a local structure alignment tool, with TM-align, a global structure alignment based method. While G-Losa predicts drug-like ligands’ positions in single-chain protein targets more precisely than TM-align, the overall success rate of TM-align is better.[37]
However, as algorithmic improvements and computer performance have erased purely technical deficiencies in older approaches, it has become clear that there is no one universal criterion for the 'optimal' structural alignment. TM-align, for instance, is particularly robust in quantifying comparisons between sets of proteins with great disparities in sequence lengths, but it only indirectly captures hydrogen bonding or secondary structure order conservation which might be better metrics for alignment of evolutionarily related proteins. Thus recent developments have focused on optimizing particular attributes such as speed, quantification of scores, correlation to alternative gold standards, or tolerance of imperfection in structural data or ab initio structural models. An alternative methodology that is gaining popularity is to use the consensus of various methods to ascertain proteins structural similarities.[38]
RNA structural alignment
Structural alignment techniques have traditionally been applied exclusively to proteins, as the primary biological macromolecules that assume characteristic three-dimensional structures. However, large RNA molecules also form characteristic tertiary structures, which are mediated primarily by hydrogen bonds formed between base pairs as well as base stacking. Functionally similar noncoding RNA molecules can be especially difficult to extract from genomics data because structure is more strongly conserved than sequence in RNA as well as in proteins,[40] and the more limited alphabet of RNA decreases the information content of any given nucleotide at any given position.
However, because of the increasing interest in RNA structures and because of the growth of the number of experimentally determined 3D RNA structures, few RNA structure similarity methods have been developed recently. One of those methods is, e.g., SETTER[41] which decomposes each RNA structure into smaller parts called general secondary structure units (GSSUs). GSSUs are subsequently aligned and these partial alignments are merged into the final RNA structure alignment and scored. The method has been implemented into the SETTER webserver.[42]
A recent method for pairwise structural alignment of RNA sequences with low sequence identity has been published and implemented in the program FOLDALIGN.[43] However, this method is not truly analogous to protein structural alignment techniques because it computationally predicts the structures of the RNA input sequences rather than requiring experimentally determined structures as input. Although computational prediction of the protein folding process has not been particularly successful to date, RNA structures without pseudoknots can often be sensibly predicted using free energy-based scoring methods that account for base pairing and stacking.[44]
Software
Choosing a software tool for structural alignment can be a challenge due to the large variety of available packages that differ significantly in methodology and reliability. A partial solution to this problem was presented in [38] and made publicly accessible through the ProCKSI webserver. A more complete list of currently available and freely distributed structural alignment software can be found in structural alignment software.
Properties of some structural alignment servers and software packages are summarized and tested with examples at Structural Alignment Tools in Proteopedia.Org.
See also
References
- Kryshtafovych A, Monastyrskyy B, Fidelis K (2016). "CASP11 statistics and the prediction center evaluation system. \". Proteins. 84 (Suppl 1): (Suppl 1):15–19. doi:10.1002/prot.25005. PMC 5479680. PMID 26857434.
- Lars Malmström Michael Riffle; Charlie EM Strauss; Dylan Chivian; Trisha N Davis; Richard Bonneau; David Baker (2007). "Superfamily Assignments for the Yeast Proteome through Integration of Structure Prediction with the Gene Ontology". PLOS Biol. 5 (4): e76corresponding author1, 2. doi:10.1371/journal.pbio.0050076. PMC 1828141. PMID 17373854.
- David E. Kim; Dylan Chivian; David Baker (2004). "Protein structure prediction and analysis using the Robetta server". Nucleic Acids Research. 32(Web Server issue): W526–W531 (Web Server issue): W526–W531. doi:10.1093/nar/gkh468. PMC 441606. PMID 15215442.
- Zhang Y, Skolnick J (2005). "The protein structure prediction problem could be solved using the current PDB library". Proc Natl Acad Sci USA. 102 (4): 1029–34. Bibcode:2005PNAS..102.1029Z. doi:10.1073/pnas.0407152101. PMC 545829. PMID 15653774.
- Zemla A. (2003). "LGA — A Method for Finding 3-D Similarities in Protein Structures". Nucleic Acids Research. 31 (13): 3370–3374. doi:10.1093/nar/gkg571. PMC 168977. PMID 12824330.
- Godzik A (1996). "The structural alignment between two proteins: Is there a unique answer?". Protein Science. 5 (7): 1325–38. doi:10.1002/pro.5560050711. PMC 2143456. PMID 8819165.
- Martin ACR (1982). "Rapid Comparison of Protein Structures". Acta Crystallogr A. 38 (6): 871–873. Bibcode:1982AcCrA..38..871M. doi:10.1107/S0567739482001806.
- Theobald DL, Wuttke DS (2006). "Empirical Bayes hierarchical models for regularizing maximum likelihood estimation in the matrix Gaussian Procrustes problem". Proceedings of the National Academy of Sciences. 103 (49): 18521–18527. Bibcode:2006PNAS..10318521T. doi:10.1073/pnas.0508445103. PMC 1664551. PMID 17130458.
- Theobald DL, Wuttke DS (2006). "THESEUS: Maximum likelihood superpositioning and analysis of macromolecular structures". Bioinformatics. 22 (17): 2171–2172. doi:10.1093/bioinformatics/btl332. PMC 2584349. PMID 16777907.
- Diederichs K. (1995). "Structural superposition of proteins with unknown alignment and detection of topological similarity using a six-dimensional search algorithm". Proteins. 23 (2): 187–95. doi:10.1002/prot.340230208. PMID 8592700. S2CID 3469775.
- Maiti R, Van Domselaar GH, Zhang H, Wishart DS (2004). "SuperPose: a simple server for sophisticated structural superposition". Nucleic Acids Res. 32 (Web Server issue): W590–4. doi:10.1093/nar/gkh477. PMC 441615. PMID 15215457.
- Ortiz, AR; Strauss CE; Olmea O. (2002). "MAMMOTH (matching molecular models obtained from theory): an automated method for model comparison". Protein Science. 11 (11): 2606–2621. doi:10.1110/ps.0215902. PMC 2373724. PMID 12381844.
- Zhang Y, Skolnick J (2004). "Scoring function for automated assessment of protein structure template quality". Proteins. 57 (4): 702–710. doi:10.1002/prot.20264. PMID 15476259. S2CID 7954787.
- Siew N, Elofsson A, Rychlewsk L, Fischer D (2000). "MaxSub: an automated measure for the assessment of protein structure prediction quality". Bioinformatics. 16 (9): 776–85. doi:10.1093/bioinformatics/16.9.776. PMID 11108700.
- Poleksic A (2009). "Algorithms for optimal protein structure alignment". Bioinformatics. 25 (21): 2751–2756. doi:10.1093/bioinformatics/btp530. PMID 19734152.
- Lathrop RH. (1994). "The protein threading problem with sequence amino acid interaction preferences is NP-complete". Protein Eng. 7 (9): 1059–68. CiteSeerX 10.1.1.367.9081. doi:10.1093/protein/7.9.1059. PMID 7831276.
- Wang L, Jiang T (1994). "On the complexity of multiple sequence alignment". Journal of Computational Biology. 1 (4): 337–48. CiteSeerX 10.1.1.408.894. doi:10.1089/cmb.1994.1.337. PMID 8790475.
- Kolodny R, Linial N (2004). "Approximate protein structural alignment in polynomial time". PNAS. 101 (33): 12201–12206. doi:10.1073/pnas.0404383101. PMC 514457. PMID 15304646.
- Martinez L, Andreani, R, Martinez, JM. (2007). "Convergent algorithms for protein structural alignment". BMC Bioinformatics. 8: 306. doi:10.1186/1471-2105-8-306. PMC 1995224. PMID 17714583.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Mount DM. (2004). Bioinformatics: Sequence and Genome Analysis 2nd ed. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY ISBN 0879697121
- Holm L, Sander C (1996). "Mapping the protein universe". Science. 273 (5275): 595–603. Bibcode:1996Sci...273..595H. doi:10.1126/science.273.5275.595. PMID 8662544. S2CID 7509134.
- Shindyalov, I.N.; Bourne P.E. (1998). "Protein structure alignment by incremental combinatorial extension (CE) of the optimal path". Protein Engineering. 11 (9): 739–747. doi:10.1093/protein/11.9.739. PMID 9796821.
- Prlic A, Bliven S, Rose PW, Bluhm WF, Bizon C, Godzik A, Bourne PE (2010). "Pre-calculated protein structure alignments at the RCSB PDB website". Bioinformatics. 26 (23): 2983–2985. doi:10.1093/bioinformatics/btq572. PMC 3003546. PMID 20937596.
- Pin-Hao Chi; Bin Pang; Dmitry Korkin; Chi-Ren Shyu (2009). "Efficient SCOP-fold classification and retrieval using index-based protein substructure alignments". Bioinformatics. 25 (19): 2559–2565. doi:10.1093/bioinformatics/btp474. PMID 19667079.
- Sara Cheek; Yuan Qi; Sri Krishna; Lisa N Kinch; Nick V Grishin (2004). "SCOPmap: Automated assignment of protein structures to evolutionary superfamilies". BMC Bioinformatics. 5 (197): 197. doi:10.1186/1471-2105-5-197. PMC 544345. PMID 15598351.
- Kai Wang; Ram Samudrala (2005). "FSSA: a novel method for identifying functional signatures from structural alignments". Bioinformatics. 21 (13): 2969–2977. doi:10.1093/bioinformatics/bti471. PMID 15860561.
- Taylor WR, Flores TP, Orengo CA (1994). "Multiple protein structure alignment". Protein Sci. 3 (10): 1858–70. doi:10.1002/pro.5560031025. PMC 2142613. PMID 7849601.
- Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM (1997). "CATH: A hierarchical classification of protein domain structures". Structure. 5 (8): 1093–1108. doi:10.1016/S0969-2126(97)00260-8. PMID 9309224.
- Zhang Y, Skolnick J (2005). "TM-align: A protein structure alignment algorithm based on the TM-score". Nucleic Acids Research. 33 (7): 2302–2309. doi:10.1093/nar/gki524. PMC 1084323. PMID 15849316.
- Stefano Angaran; Mary Ellen Bock; Claudio Garutti; Concettina Guerra1 (2009). "MolLoc: a web tool for the local structural alignment of molecular surfaces". Nucleic Acids Research. 37 (Web Server issue): W565–70. doi:10.1093/nar/gkp405. PMC 2703929. PMID 19465382.
- Gaëlle Debret; Arnaud Martel; Philippe Cuniasse (2009). "RASMOT-3D PRO: a 3D motif search webserver". Nucleic Acids Research. 37 (Web Server issue): W459–64. doi:10.1093/nar/gkp304. PMC 2703991. PMID 19417073.
- Alexandra Shulman-Peleg; Maxim Shatsky; Ruth Nussinov; Haim J. Wolfson (2008). "MultiBind and MAPPIS: webservers for multiple alignment of protein 3D-binding sites and their interactions". Nucleic Acids Research. 36 (Web Server issue): W260–4. doi:10.1093/nar/gkn185. PMC 2447750. PMID 18467424.
- Alexandra Shulman-Peleg; Maxim Shatsky; Ruth Nussinov; Haim J Wolfson (2007). "Spatial chemical conservation of hot spot interactions in protein-protein complexes". BMC Biology. 5 (43): 43. doi:10.1186/1741-7007-5-43. PMC 2231411. PMID 17925020.
- Gabriele Ausiello; Pier Federico Gherardini; Paolo Marcatili; Anna Tramontano; Allegra Via; Manuela Helmer-Citterich (2008). "FunClust: a web server for the identification of structural motifs in a set of non-homologous protein structures". BMC Biology. 9 (Suppl 2): S2. doi:10.1186/1471-2105-9-S2-S2. PMC 2323665. PMID 18387204.
- Janez Konc; Dušanka Janežič (2010). "ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment". Bioinformatics. 26 (9): 1160–1168. doi:10.1093/bioinformatics/btq100. PMC 2859123. PMID 20305268.
- Hui Sun Lee; Wonpil Im (2012). "Identification of Ligand Templates using Local Structure Alignment for Structure-Based Drug Design". Journal of Chemical Information and Modeling. 52 (10): 2784–2795. doi:10.1021/ci300178e. PMC 3478504. PMID 22978550.
- Hui Sun Lee; Wonpil Im (2013). "Ligand Binding Site Detection by Local Structure Alignment and Its Performance Complementarity". Journal of Chemical Information and Modeling. 53 (9): 2462–2470. doi:10.1021/ci4003602. PMC 3821077. PMID 23957286.
- Barthel D., Hirst J.D., Blazewicz J., Burke E.K. and Krasnogor N. (2007). "ProCKSI: a decision support system for Protein (Structure) Comparison, Knowledge, Similarity and Information". BMC Bioinformatics. 8: 416. doi:10.1186/1471-2105-8-416. PMC 2222653. PMID 17963510.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Sippl, M.; Wiederstein, M. (2012). "Detection of spatial correlations in protein structures and molecular complexes". Structure. 20 (4): 718–728. doi:10.1016/j.str.2012.01.024. PMC 3320710. PMID 22483118.
- Torarinsson E, Sawera M, Havgaard JH, Fredholm M, Gorodkin J (2006). "Thousands of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA structure". Genome Res. 16 (7): 885–9. doi:10.1101/gr.5226606. PMC 1484455. PMID 16751343.
- Hoksza D, Svozil D (2012). "Efficient RNA pairwise structure comparison by SETTER method". Bioinformatics. 28 (14): 1858–1864. doi:10.1093/bioinformatics/bts301. PMID 22611129.
- Cech P, Svozil D, Hoksza D (2012). "SETTER: web server for RNA structure comparison". Nucleic Acids Research. 40 (W1): W42–W48. doi:10.1093/nar/gks560. PMC 3394248. PMID 22693209.
- Havgaard JH, Lyngso RB, Stormo GD, Gorodkin J (2005). "Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%". Bioinformatics. 21 (9): 1815–24. doi:10.1093/bioinformatics/bti279. PMID 15657094.
- Mathews DH, Turner DH (2006). "Prediction of RNA secondary structure by free energy minimization". Curr Opin Struct Biol. 16 (3): 270–8. doi:10.1016/j.sbi.2006.05.010. PMID 16713706.
Further reading
- Bourne PE, Shindyalov IN. (2003): Structure Comparison and Alignment. In: Bourne, P.E., Weissig, H. (Eds): Structural Bioinformatics. Hoboken NJ: Wiley-Liss. ISBN 0-471-20200-2
- Yuan X, Bystroff C. (2004) "Non-sequential Structure-based Alignments Reveal Topology-independent Core Packing Arrangements in Proteins", Bioinformatics. Nov 5, 2004
- Jung J, Lee B (2000). "Protein structure alignment using environmental profiles". Protein Eng. 13 (8): 535–543. doi:10.1093/protein/13.8.535. PMID 10964982.
- Ye Y, Godzik A (2005). "Multiple flexible structure alignment using partial order graphs". Bioinformatics. 21 (10): 2362–2369. doi:10.1093/bioinformatics/bti353. PMID 15746292.
- Sippl M, Wiederstein M (2008). "A note on difficult structure alignment problems". Bioinformatics. 24 (3): 426–427. doi:10.1093/bioinformatics/btm622. PMID 18174182.