Polycystic kidney disease 3 (autosomal dominant)
Polycystic kidney disease 3 (autosomal dominant) is a protein that in humans is encoded by the PKD3 gene.[1]
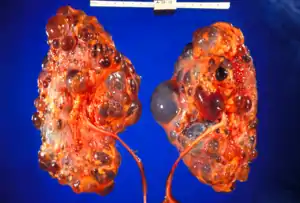
Polycystic kidney disease (ADPKD) is a life threatening hereditary disorder; it is characterized by the development of fluid-filled cyst formation and expansion of the kidney and other organs.[2] It is an autosomal dominant disease, and it is the most common hereditary disorders with a rate of occurrence of approximately 1 in 1000.[3]
Characteristics
ADPKD is an autosomal dominant disease, it contains 3 types of mutation: PKD1 (16 Chr), PKD2 (4 Chr) PKD3 (11 Chr, this gene). Mutations in the 3 different genes PKD1, PKD2 and PKD3 cause a very similar disorder of the autosomal dominant form of polycystic kidney disease (ADPKD).[4] The PKD3 gene is located on chromosome number 11q12.3; the phenotype MIM number is 600666.
PKD3 it is the result of a mutation in the GANAB gene. The GANAB gene codes the catalytic alpha subunit of glucosidase II and noncatalytic beta subunit; the glycosyl hydrolase 31 families of proteins. The heterodimeric glucosidase II enzyme has an important function in protein folding and catalyzes the hydrolysis of glucose residues. In the endoplasmic reticulum, the quality control is done by cutting the residues of glucose from the immature glycoproteins. The rise in the expression of the protein leads to the lung tumor tissue. Also, any mutation in this gene causes autosomal dominant polycystic liver disease. [7]
PKD3 occurs in adults and it sometimes shows symptoms in children. It is known as 'polycystic kidneys'. Polycystic kidney disease (PKD3) is an autosomal dominant inheritance that leads to renal cysts. It is related to the liver cysts that sometimes causes organ dysfunction. It is usually detected in middle to late aged individuals developing severe cysts in kiddy and liver. However, the renal disease is mild and very few patients have hypertension, it does not happen regularly. Unlike the liver disease where it develops a wide spectrum of severity; some have no cyst, while others have severe liver involvement polycystic kidney disease.[2]
Clinical features
In an analysis of 20 patients from 9 unrelated families develop polycystic kidney disease with heterozygous mutations in the GANAB gene. 5 of the mutations from the GANAB gene were predicted to result in a short protein (frameshift, nonsense, or splicing), and 3 missense mutations. 7 of the families had a diagnosis of PKD, while the other 2 families had a diagnosis of polycystic liver disease (PCLD). Most of the patients developed during adulthood, except one showed symptoms at the age of 9. The recordings indicate that the renal disease was insignificant. Also, very few of them had high blood pressure. Renal imaging has also been done, which present variable numbers of multiple cysts (from less than 10 to more than 40).Those suffering from PCLD, some had no cyst while others develop severe disease, requiring surgery. Therefore, this shows that GANAB-related PKD and PCLD are not necessarily separate diseases, but there was significant phenotypic overlap between the 2, and that the PKD3 is responsible for this disease.[3]
Treatment
There is no specific treatment for PKD3. However, a lot of research is going on to mimic the symptoms. Regular monitoring and following up on the complications can assist in maintaining the health and extend a person's life. Recent research shows that the treatment with protein kinase C (PKC) inhibitor may help to mimic the increase of PKD3, by RacV12 and aluminum fluoride.
Mechanism
- Both Galpha (12/13) and Rac are important components in the signal transduction pathways that mediate bombesin receptor induced PKD3 activation.[5]
- The addition aluminum fluoride to COS-7 cells cotransfected with PKD3 and Gapha (13/12) leads to PKD3 activation; also it is related with a transient plasma membrane translocation of cytosolic PKD3.[5]
- The catalytic domain of PKD3 can localize to the nucleus when expressed without the kinase domain.[6] Moreover, the catalytic activation of PKD3 in response to RacV12, Galpha (13/12) signalling, bombesin correlated with Ser-731/735 phosphorylation in the activation loop of this enzyme.[5]
References
- "Entrez Gene: Polycystic kidney disease 3 (autosomal dominant)".
- Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, et al. (2016). "Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease". American Journal of Human Genetics. 98 (6): 1193–207. doi:10.1016/j.ajhg.2016.05.004. PMC 4908191. PMID 27259053.
- Paul BM, Consugar MB, Ryan Lee M, Sundsbak JL, Heyer CM, Rossetti S, Kubly VJ, Hopp K, Torres VE, Coto E, Clementi M, Bogdanova N, de Almeida E, Bichet DG, Harris PC (2014). "Evidence of a third ADPKD locus is not supported by re-analysis of designated PKD3 families". Kidney International. 85 (2): 383–92. doi:10.1038/ki.2013.227. PMC 3883953. PMID 23760289.
- Koptides M, Deltas CC (2000). "Autosomal dominant polycystic kidney disease: molecular genetics and molecular pathogenesis". Human Genetics. 107 (2): 115–26. doi:10.1007/s004390000347. PMID 11030408. S2CID 11284208.
- Yuan J, Rey O, Rozengurt E (2006). "Activation of protein kinase D3 by signaling through Rac and the alpha subunits of the heterotrimeric G proteins G12 and G13". Cellular Signalling. 18 (7): 1051–62. doi:10.1016/j.cellsig.2005.08.017. PMID 16198087.
- Papazyan R, Rozengurt E, Rey O (2006). "The C-terminal tail of protein kinase D2 and protein kinase D3 regulates their intracellular distribution". Biochemical and Biophysical Research Communications. 342 (3): 685–9. doi:10.1016/j.bbrc.2006.02.013. PMID 16494840.
[7] GANAB glucosidase II alpha subunit Homo sapiens (human). Gene ID:23193, 2016.
Further reading
- Chen J, Deng F, Singh SV, Wang QJ (May 2008). "Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2". Cancer Research. 68 (10): 3844–53. doi:10.1158/0008-5472.CAN-07-5156. PMID 18483269.
- Potter EL (1972). Normal and Abnormal Development of the Kidney. Chicago: Year Book Medical Publishers. ISBN 978-0-8151-6763-1.