PDE3 inhibitor
A PDE3 inhibitor is a drug which inhibits the action of the phosphodiesterase enzyme PDE3. They are used for the therapy of acute heart failure and cardiogenic shock.
Medical uses
Cardiac
Amrinone, milrinone and enoximone are used clinically for short-term treatment of cardiac failure in the presence of cardiogenic shock.[1]
PDE3 inhibitors are indicated as inotropics for the therapy of acute heart failure if catecholamines are ineffective.[2] Well controlled studies have shown that these drugs generally increase mortality,[3] when used for the therapy of acute heart failure, so they have to be applied under close observation.[1]
Peripheral artery disease
Cilostazol is used for the treatment of intermittent claudication. This drug has a much weaker positive inotropic effect than those drugs used for the therapy of acute heart failure, and lacks significant adverse cardiac effects.[4]
Contraindications
Cardiac
Contraindications are severe obstructive cardiomyopathy, hypovolemia, tachycardia, and ventricular aneurysm. Breast feeding is prohibited during treatment.[1]
Adverse effects
Cardiac
The most important adverse effects when used for the therapy of acute heart failure are arrhythmia, thrombocytopenia and increased transaminase levels.[1][2]
Types
Approved PDE3 inhibitors include the following:
- amrinone
- cilostazol
- milrinone
- enoximone
- pimobendan (approved for human use in Japan,[5] approved for dogs elsewhere)
Mechanism of action
PDE3 inhibitors are a type of phosphodiesterase inhibitors. Inhibition of the PDE isoenzyme 3 leads to an increase of intracellular concentrations of the second messenger cyclic adenosine monophosphate (cAMP). cAMP mediates the phosphorylation of protein kinases, which in turn activates cardiac calcium channels. An increased calcium influx from the sarcoplasmic reticulum (SR) during phase 2 (the plateau phase) of the cardiac action potential leads to a positive inotropic effect of PDE3 inhibitors: they increase the force of cardiac contraction. Increased reflux of calcium into the SR following the plateau phase is responsible for their positive lusitropic effect: they increase relaxation speed. Additionally, PDE3 inhibitors act as vasodilators.[1][2]
Chemical properties
First generation PDE3 inhibitors
Recognition that the knowledge about PDE could be used to develop drugs that were PDE inhibitors led to extensive research. Most studies used analogues of the nucleotide substrates or derivatives of natural product inhibitors such as xanthine (e.g. theophylline) and papaverine.[6][7]
The active site of PDE3 can be considered as a summary of ideas about receptor topography resulting from the first generation inhibitors. The model of the Wells et al. version as cited in Erhardt and Chou (1991) includes the following:
- A phosphate binding area
- A lipophilic area that accommodates the non-polar side of the ribose moiety
- A pyrimidine binding site
- An imidazole binding site portion of the pyrimidine binding site
- A sterically hindered site
- An area with bulk tolerance[6]
Second generation PDE3 inhibitors
Since selective PDE3 inhibitors were recognised to be cardiotonic drugs there has been great interest in developing new drugs in this category. A large number of heterocyclic compounds have been synthesized during related research. These compounds constitute a second generation of PDE inhibitors. Although they have been directed mostly at PDE3, they present significant structure-activity relationship for the PDEs in general.[6]
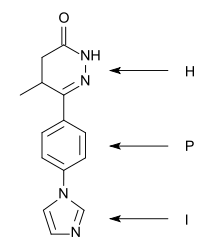
A "heterocycle-phenyl-imidazole" (H-P-I) pattern has been considered to be necessary for positive inotropic activity in cardiac muscle and many second generation inhibitors fit this pattern.[6]
The heterocycle region: Within each heterocycle there is the presence of a dipole and an adjacent acid proton (an amide function). These atoms are believed to mimic the electrophilic center in the phosphate group in cAMP and are confirmed as the primary site of binding. The heterocycle is a transition state analogue inhibitor of PDE. Alkyl groups, limited to either methyl or ethyl, on the heterocyclic ring usually enhance potency, with occasional exceptions.[6][7]
The phenyl region: It seems that an electron rich centre, such as phenyl, needs to be present. The beneficial effects of small alkyl groups on the heterocycle could be to twist the central ring away from exact coplanarity with the heterocyclic ring. There is a similar twist in cAMP and there is general agreement that high affinity PDE3 inhibitors should adopt an energetically favoured planar conformation that mimics the anti conformation of cAMP.[6][7]
The imidazole region: Various substituents have been placed at the para-position of the central phenyl ring. They are electron rich moieties and apparently a positively charged moiety cannot be tolerated in this region of the PDE receptor. There is general agreement about this inhibitor potency: lactam ≥ alkyl-CONH- ≥ imidazoyl = pyridine in place of the central phenyl with its nitrogen in the analogous 4 position ≥ alkyl-S- > simple ether > halide = amine > imidazolium (which is totally inactive).[6]
Identification of features common to the most selective inhibitors has led to a "five-point model" with:
Examples of selective PDE3 inhibitors
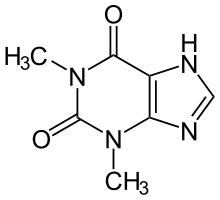 Theophylline, a non-selective inhibitor |
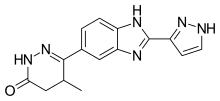 Meribendan, a highly selective inhibitor |
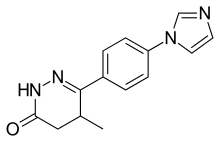
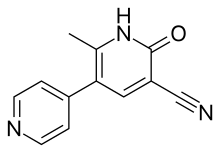
Theophylline is a non-selective agent. In contrast, meribendan is a highly selective inhibitor.[7]
Also, meribendan has a higher level of selectivity in comparison with the parent compound CI-930 because, beside the basic nitrogen adjacent to the lactam moiety it possesses another basic nitrogen (benzimidazole ring), opposite to the primary binding site.[7]
Research
RPL-554 is an analog of trequinsin, and like trequinsin, is a dual inhibitor of the phosphodiesterase enzymes PDE-3 and PDE-4.[8] As of October 2015, inhaled RPL-554 delivered via a nebulizer was in development for COPD and had been studied in asthma.[9]
References
- Mutschler, Ernst; Schäfer-Korting, Monika (1997). Arzneimittelwirkungen (in German). Stuttgart: Wissenschaftliche Verlagsgesellschaft. pp. 454–455, 496.
- Forth; Henschler; Rummel (2002). Allgemeine und spezielle Pharmakologie und Toxikologie (in German). Munich. p. 457.
{{cite book}}: CS1 maint: location missing publisher (link) - "Approval of Cilostazol". FDA. 25 September 2006. Archived from the original on 27 April 2007.
- Yan, Chen. "Regulation of Phosphodiesterase 3 and Inducible cAMP Early Repressor in the Heart". Circulation Research. Retrieved 23 January 2016.
- Mizuno, Masashi; Yamano, Shigeki; Chimura, Shuichi; Hirakawa, Atsushi; Takusagawa, Yoshimi; Sawada, Tamotsu; Maetani, Shigeki; Takahashi, Arane; Mizuno, Takeshi; Harada, Kayoko; Shinoda, Asako (20 January 2017). "Efficacy of pimobendan on survival and reoccurrence of pulmonary edema in canine congestive heart failure". The Journal of Veterinary Medical Science. 79 (1): 29–34. doi:10.1292/jvms.16-0069. PMC 5289233. PMID 27644192.
- Erhardt, Paul W.; Chou, Yuo-Ling (January 1991). "A topographical model for the c-AMP phosphodiesterase III active site". Life Sciences. 49 (8): 553–568. doi:10.1016/0024-3205(91)90254-9. PMID 1650876.
- Fossa, Paola; Boggia, Raffaella; Mosti, Luisa (1998). "Toward the identification of the cardiac cGMP inhibited-phosphodiesterase catalytic site". Journal of Computer-Aided Molecular Design. 12 (4): 361–372. Bibcode:1998JCAMD..12..361F. doi:10.1023/a:1007928412086. PMID 9777494. S2CID 34807211.
- Boswell-Smith, Victoria; Spina, Domenico; Oxford, Alec W.; Comer, Mike B.; Seeds, Esther A.; Page, Clive P. (August 2006). "The pharmacology of two novel long-acting phosphodiesterase 3/4 inhibitors, RPL554 [9,10-dimethoxy-2(2,4,6-trimethylphenylimino)-3-(n-carbamoyl-2-aminoethyl)-3,4,6,7-tetrahydro-2H-pyrimido[6,1-a]isoquinolin-4-one] and RPL565 [6,7-dihydro-2-(2,6-diisopropylphenoxy)-9,10-dimethoxy-4H-pyrimido[6,1-a]isoquinolin-4-one]". The Journal of Pharmacology and Experimental Therapeutics. 318 (2): 840–848. doi:10.1124/jpet.105.099192. PMID 16682455. S2CID 15490792.
- Taylor, Nick Paul (1 October 2015). "Verona sets sights on PhIIb after COPD drug comes through early trial". FierceBiotech. Archived from the original on 7 March 2016. Retrieved 2 March 2016.