Phage-assisted continuous evolution
Phage-assisted continuous evolution (PACE) is a phage-based technique for the automated directed evolution of proteins. It relies on relating the desired activity of a target protein with the fitness of an infectious bacteriophage which carries the protein's corresponding gene. Proteins with greater desired activity hence confer greater infectivity to their carrier phage. More infectious phage propagate more effectively, selecting for advantageous mutations. Genetic variation is generated using error-prone polymerases on the phage vectors, and over time the protein accumulates beneficial mutations. This technique is notable for performing hundreds of rounds of selection with minimal human intervention.
Principle
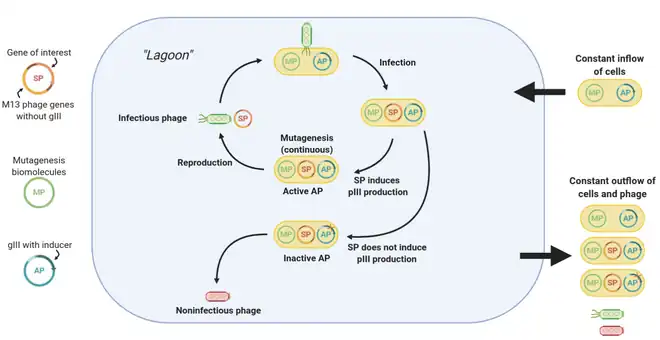
The central component of PACE is a fixed-volume vessel known as the “lagoon”. The lagoon contains M13 bacteriophage vectors carrying the gene of interest (known as the selection plasmid, or SP), as well as host E. coli cells that allow the phage to replicate. The lagoon is constantly diluted via the addition and draining of liquid media containing E. coli cells. The liquid flow rate is set such that the dilution rate is faster than the rate of E. coli reproduction but slower than the rate of phage reproduction. Hence, a fresh supply of E. coli cells is constantly present in the lagoon, but phage can only be retained via sufficiently fast replication.[1]
Phage replication requires E. coli infection, which, for M13 phage, relies on protein III (pIII).[2] When using PACE, the phage vectors lack the gene to produce pIII. Instead, the production of pIII is tied with the activity of the protein of interest via a mechanism that varies per use case, oftentimes involves an extra plasmid containing the pIII-expressing gene III (gIII) known as the accessory plasmid, or AP. Notably, production of infectious phage scales with the production of pIII.[3] Hence, the better the activity of the protein, the higher the rate of pIII production, and the more infectious phage are generated for that particular gene.
Using error-prone polymerases (encoded on the mutagenesis plasmid, or MP), genetic variation is introduced into the protein gene portion of the phage vectors. Due to the selective pressures applied by the constant draining of the lagoon, only phages that can replicate fast enough can be retained in the lagoon, so over time beneficial mutations accumulate in phage replicating in the lagoon. In this manner, rounds of evolution are continuously performed, allowing hundreds of rounds to elapse with little human intervention.[1]
Applications
Polymerase promoter specificity
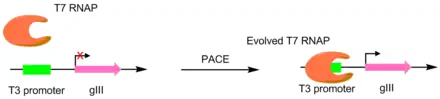
In the initial paper pioneering this technique, T7 RNA polymerases were evolved to recognize different promoters, such as the T3 or SP6 promoters.[4] This was done by making the target promoter the sole promoter for gIII.[5] Hence, mutant polymerases with greater specificity for the desired promoter caused greater pIII production. This resulted in polymerases with ~3-4 orders of magnitude greater activity for the target promoter than the original T3 promoter.[4] While this original PACE system only performed positive selection, a variant was developed that allowed for negative selection as well. This is done by linking undesired activity to the production of non-functional pIII, which decreases the amount of infectious phage made.[6]
Protease substrate specificity
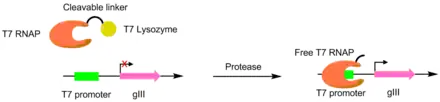
Proteases have been evolved to cut different peptides using PACE. In these systems, the desired protease cut site is used to link a T7 RNA polymerase and a T7 lysozyme. The T7 lysozyme prevents the T7 polymerase from transcribing gIII. When the peptide linker is cleaved, the T7 polymerase is activated, allowing for the transcription of the pIII gene. This method was used to create a TEV protease with a significantly different peptide substrate.[6][7]
Orthogonal Aminoacyl-tRNA Synthetases
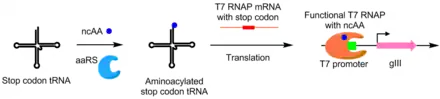
Using PACE, aminoacyl-tRNA synthetases (aaRSs) were evolved for noncanonical amino acids as well. Activity of an aaRS is linked to pIII production by the addition of a TAG stop codon in the middle of gIII. Synthetases that aminoacylate the TAG codon's suppressor tRNA prevents stop codon activity, allowing for production of functional pIII. Using this system, aaRSs were evolved that utilize non-canonical amino acids p-nitro-phenyalanine, iodophenylalanine, and Boc-lysine.[8]
Protein-Protein Interactions
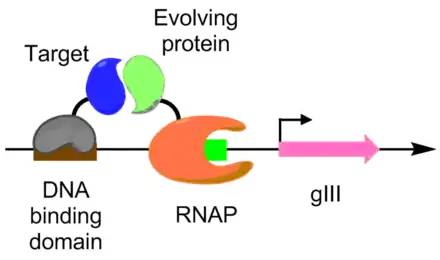
Protein-protein interactions have been evolved using PACE as well. Under this scheme, the target protein is fused with a DNA binding protein, which binds to a target sequence placed upstream of the gIII promoter. The protein undergoing evolution is fused with an RNA polymerase. The better the protein-protein interaction, the more transcription of pIII occurs, allowing the evolution of the protein-protein interaction under PACE conditions.[6] This method was used to evolve Bacillus thuringiensis endotoxin variants that can overcome insect toxin resistance.[6][9]
Base Editors
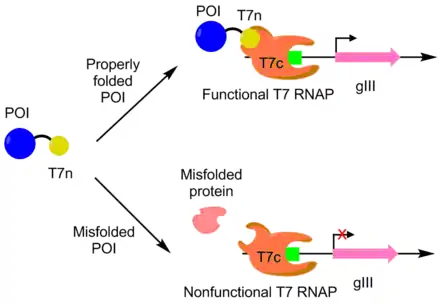
PACE was used to evolve APOBEC1 for greater soluble expression. APOBEC1 is a cytidine deaminase that has found use in base editors to catalyze the single nucleotide edit C-->T.[10] In E. coli, APOBEC1 usually falls out of solution into the insoluble fraction.[11] To evolve APOBEC1 for better soluble expression, the N-terminus of a T7 polymerase was fused to APOBEC1, with the remaining portion of the polymerase separately expressed. The T7 polymerase can only function when the N-terminus portion can bind to the rest of the polymerase. Since APOBEC1 must be properly folded for the N-terminus portion to be exposed properly, T7 polymerase activity is correlated to APOBEC1 folding. As follows, pIII transcription and production is linked with APOBEC1 soluble expression via the T7 polymerase. Using this approach, the soluble expression of APOBEC1 was increased by 4 fold with no change in function.[7][9]
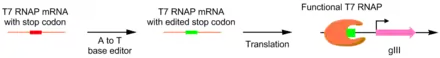
PACE was also used to create a more catalytically active deoxyadenosine deaminase. Deoxyadenosine deaminase is used in base editors to perform the single nucleotide edit A-->T. This was done by placing adenosine-containing stop codons in the gene for T7 polymerase. If the base editor is able to correct the error, functional T7 polymerase is produced, allowing production of pIII. Using this system, they evolved a deoxyadenosine deaminase with 590 fold activity compared to wild type.[12]
References
- Esvelt, K.; Carlson, J.; Liu, D.R. (2011). "A system for the continuous directed evolution of biomolecules". Nature. 472 (7344): 499–503. Bibcode:2011Natur.472..499E. doi:10.1038/nature09929. PMC 3084352. PMID 21478873.
- Riechmann, L.; Holliger, P. (1997). "The C-terminal domain of TolA is the coreceptor for filamentous phage infection of E. coli". Cell. 90 (2): 351–360. doi:10.1016/s0092-8674(00)80342-6. PMID 9244308.
- Rakonjac, J.; Model, P. (1998). "Roles of pIII in filamentous phage assembly". J. Mol. Biol. 282 (1): 25–41. doi:10.1006/jmbi.1998.2006. PMID 9733639.
- Lane, M.D.; Seelig, B. (2014). "Advances in the directed evolution of proteins". Curr. Opin. Chem. Biol. 22: 129–136. doi:10.1016/j.cbpa.2014.09.013. PMC 4253873. PMID 25309990.
- Lemire, S.; Yehl, K.M.; Lu, T.K. (2018). "Phage-Based Applications in Synthetic Biology". Annu. Rev. Virol. 5 (1): 453–476. doi:10.1146/annurev-virology-092917-043544. PMC 6953747. PMID 30001182.
- Brödel, A.K.; Isalan, M.; Jaramillo, A. (2018). "Engineering of biomolecules by bacteriophage directed evolution". Curr. Opin. Biotech. 51: 32–38. doi:10.1016/j.copbio.2017.11.004. PMID 29175708.
- Kim, J.Y.; Yoo, H.W.; Lee, P.G.; Lee, S.G.; Seo, J.H.; Kim, B.G. (2019). "In vivo Protein Evolution, Next Generation Protein Engineering Strategy: from Random Approach to Target-specific Approach". Biotechnol. Bioproc. E. 24: 85–94. doi:10.1007/s12257-018-0394-2. S2CID 91687131.
- Vargas-Rodriguez, O.; Sevostyanova, A.; Söll, D.; Crnković, A. (2018). "Upgrading aminoacyl-tRNA synthetases for genetic code expansion". Curr. Opin. Chem. Biol. 46C: 115–122. doi:10.1016/j.cbpa.2018.07.011. PMC 7083171. PMID 30056281.
- Simon, A.J.; d'Oelsnitz, S.; Ellington, A.D. (2018). "Synthetic Evolution". Nat. Biotechnol. 37 (7): 730–743. doi:10.1038/s41587-019-0157-4. PMID 31209374. S2CID 189927244.
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. (2017). "Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage". Nature. 551 (7681): 464–471. doi:10.1038/nature24644. PMC 5726555. PMID 29160308.
- Wang, T.; Badran, A.H.; Huang, T.P.; Liu, D.R. (2018). "Continuous directed evolution of proteins with improved soluble expression". Nat. Chem. Biol. 14 (10): 972–980. doi:10.1038/s41589-018-0121-5. PMC 6143403. PMID 30127387.
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; Doudna, J.A.; Liu, D.R. (2020). "Phage-Assisted Evolution of an Adenine Base Editor with Enhanced Cas Domain Compatibility and Activity". Nat. Biotechnol. 38 (7): 883–891. doi:10.1038/s41587-020-0453-z. PMC 7357821. PMID 32433547.