Nernst equation
In electrochemistry, the Nernst equation is a chemical thermodynamical relationship that permits the calculation of the reduction potential of a reaction (half-cell or full cell reaction) from the standard electrode potential, absolute temperature, the number of electrons involved in the redox reaction, and activities (often approximated by concentrations) of the chemical species undergoing reduction and oxidation respectively. It was named after Walther Nernst, a German physical chemist who formulated the equation.[1][2]
Expression
General form with chemical activities
When an oxidizer (Ox) accepts a number z of electrons ( e−) to be converted in its reduced form (Red), the half-reaction is expressed as:
- Ox + z e− → Red
The reaction quotient (Qr), also often called the ion activity product (IAP), is the ratio between the chemical activities (a) of the reduced form (the reductant, aRed) and the oxidized form (the oxidant, aOx). The chemical activity of a dissolved species corresponds to its true thermodynamic concentration taking into account the electrical interactions between all ions present in solution at elevated concentrations. For a given dissolved species, its chemical activity (a) is the product of its activity coefficient (γ) by its molar (mol/L solution), or molal (mol/kg water), concentration (C): a = γ C. So, if the concentration (C, also denoted here below with square brackets [ ]) of all the dissolved species of interest are sufficiently low and that their activity coefficients are close to unity, their chemical activities can be approximated by their concentrations as commonly done when simplifying, or idealizing, a reaction for didactic purposes:
![{\displaystyle Q_{r}={\frac {a_{\text{Red}}}{a_{\text{Ox}}}}={\frac {[Red]}{[Ox]}}}](../I/59ed999dc51d86c353f5c77be1376388e2fc6e27.svg)
At chemical equilibrium, the ratio Qr of the activity of the reaction product (aRed) by the reagent activity (aOx) is equal to the equilibrium constant K of the half-reaction:

The standard thermodynamics also says that the actual Gibbs free energy ΔG is related to the free energy change under standard state ΔGo
by the relationship:

where Qr is the reaction quotient and R is the ideal gas constant.
The cell potential E associated with the electrochemical reaction is defined as the decrease in Gibbs free energy per coulomb of charge transferred, which leads to the relationship

The constant F (the Faraday constant) is a unit conversion factor F = NAq, where NA is the Avogadro constant and q is the fundamental electron charge. This immediately leads to the Nernst equation, which for an electrochemical half-cell is

For a complete electrochemical reaction (full cell), the equation can be written as

where:
- Ered is the half-cell reduction potential at the temperature of interest,
- E
o
red is the standard half-cell reduction potential, - Ecell is the cell potential (electromotive force) at the temperature of interest,
- E
o
cell is the standard cell potential, - R is the universal gas constant: R = 8.31446261815324 J K−1 mol−1,
- T is the temperature in kelvins,
- z is the number of electrons transferred in the cell reaction or half-reaction,
- F is the Faraday constant, the magnitude of charge (in coulombs) per mole of electrons: F = 96485.3321233100184 C mol−1,
- Qr is the reaction quotient of the cell reaction, and
- a is the chemical activity for the relevant species, where aRed is the activity of the reduced form and aOx is the activity of the oxidized form.
Thermal voltage
At room temperature (25 °C), the thermal voltage is approximately 25.693 mV. The Nernst equation is frequently expressed in terms of base-10 logarithms (i.e., common logarithms) rather than natural logarithms, in which case it is written:


where λ = ln(10) ≈ 2.3026 and λVT ≈ 0.05916 Volt.
Form with activity coefficients and concentrations
Similarly to equilibrium constants, activities are always measured with respect to the standard state (1 mol/L for solutes, 1 atm for gases, and T = 298.15 K, i.e., 25 °C or 77 °F). The chemical activity of a species i, ai, is related to the measured concentration Ci via the relationship ai = γi Ci, where γi is the activity coefficient of the species i. Because activity coefficients tend to unity at low concentrations, or are unknown or difficult to determine at medium and high concentrations, activities in the Nernst equation are frequently replaced by simple concentrations and then, formal standard reduction potentials used.

Taking into account the activity coefficients () the Nernst equation becomes:




Where the first term including the activity coefficients () is denoted and called the formal standard reduction potential, so that can be directly expressed as a function of and the concentrations in the simplest form of the Nernst equation:


Formal standard reduction potential
When wishing to use simple concentrations in place of activities, but that the activity coefficients are far from unity and can no longer be neglected and are unknown or too difficult to determine, it can be convenient to introduce the notion of the "so-called" standard formal reduction potential () which is related to the standard reduction potential as follows:[3]

So that the Nernst equation for the half-cell reaction can be correctly formally written in terms of concentrations as:
and likewise for the full cell expression.
According to Wenzel (2020),[4] a formal reduction potential is the reduction potential that applies to a half reaction under a set of specified conditions such as, e.g., pH, ionic strength, or the concentration of complexing agents.
The formal reduction potential is often a more convenient, but conditional, form of the standard reduction potential, taking into account activity coefficients and specific conditions characteristics of the reaction medium. Therefore, its value is a conditional value, i.e., that it depends on the experimental conditions and because the ionic strength affects the activity coefficients, will vary from medium to medium.[3] Several definitions of the formal reduction potential can be found in the literature, depending on the pursued objective and the experimental constraints imposed by the studied system. The general definition of refers to its value determined when . A more particular case is when is also determined at pH 7, as e.g. for redox reactions important in biochemistry or biological systems.

Determination of the formal standard reduction potential when Cred/Cox = 1
The formal standard reduction potential can be defined as the measured reduction potential of the half-reaction at unity concentration ratio of the oxidized and reduced species (i.e., when Cred/Cox = 1) under given conditions.[5]
Indeed:
as, , when ,


- , when ,

because , and that the term is included in .


The formal reduction potential makes possible to more simply work with molar (mol/L, M) or molal (mol/kg H2O, m) concentrations in place of activities. Because molar and molal concentrations were once referred as formal concentrations, it could explain the origin of the adjective formal in the expression formal potential.
The formal potential is thus the reversible potential of an electrode at equilibrium immersed in a solution where reactants and products are at unit concentration.[6] If any small incremental change of potential causes a change in the direction of the reaction, i.e. from reduction to oxidation or vice versa, the system is close to equilibrium, reversible and is at its formal potential. When the formal potential is measured under standard conditions (i.e. the activity of each dissolved species is 1 mol/L, T = 298.15 K = 25 °C = 77 °F, Pgas = 1 bar) it becomes de facto a standard potential.[7]
According to Brown and Swift (1949):
"A formal potential is defined as the potential of a half-cell, measured against the standard hydrogen electrode, when the total concentration of each oxidation state is one formal".[8]
In this case, as for the standard reduction potentials, the concentrations of dissolved species remain equal to one molar (M) or one molal (m), and so are said to be one formal (F). So, expressing the concentration C in molarity M (1 mol/L):

The term formal concentration (F) is now largely ignored in the current literature and can be commonly assimilated to molar concentration (M), or molality (m) in case of thermodynamic calculations.[9]
The formal potential is also found half way between the two peaks in a cyclic voltammogram, where at this point the concentration of Ox (the oxidized species) and Red (the reduced species) at the electrode surface are equal.
The activity coefficients and are included in the formal potential , and because they depend on experimental conditions such as temperature, ionic strength, and pH, cannot be referred as an immutable standard potential but needs to be systematically determined for each specific set of experimental conditions.[7]


Formal reduction potentials are applied to simplify calculations of a considered system under given conditions and measurements interpretation. The experimental conditions in which they are determined and their relationship to the standard reduction potentials must be clearly described to avoid to confuse them with standard reduction potentials.
Formal standard reduction potential at pH 7
Formal standard reduction potentials () are also commonly used in biochemistry and cell biology for referring to standard reduction potentials measured at pH 7, a value closer to the pH of most physiological and intracellular fluids than the standard state pH of 0. The advantage is to defining a more appropriate redox scale better corresponding to real conditions than the standard state. Formal standard reduction potentials () allow to more easily estimate if a redox reaction supposed to occur in a metabolic process or to fuel microbial activity under some conditions is feasible or not.
While, standard reduction potentials always refer to the standard hydrogen electrode (SHE), with [ H+] = 1 M corresponding to a pH 0, and fixed arbitrarily to zero by convention, it is no longer the case at a pH of 7. Then, the reduction potential of a hydrogen electrode operating at pH 7 is -0.413 V with respect to the standard hydrogen electrode (SHE).[10]

Expression of the Nernst equation as a function of pH
The and pH of a solution are related by the Nernst equation as commonly represented by a Pourbaix diagram ( – pH plot). explicitly denotes expressed versus the standard hydrogen electrode (SHE). For a half cell equation, conventionally written as a reduction reaction (i.e., electrons accepted by an oxidant on the left side):


The half-cell standard reduction potential is given by


where is the standard Gibbs free energy change, z is the number of electrons involved, and F is the Faraday's constant. The Nernst equation relates pH and as follows:


where curly brackets indicate activities, and exponents are shown in the conventional manner. This equation is the equation of a straight line for as a function of pH with a slope of volt (pH has no units).

This equation predicts lower at higher pH values. This is observed for the reduction of O2 into H2O, or OH−, and for the reduction of H+ into H2. is then often noted as to indicate that it refers to the standard hydrogen electrode (SHE) whose = 0 by convention under standard conditions (T = 298.15 K = 25 °C = 77 F, Pgas = 1 atm (1.013 bar), concentrations = 1 M and thus pH = 0).
Main factors affecting the formal standard reduction potentials
The main factor affecting the formal reduction potentials in biochemical or biological processes is most often the pH. To determine approximate values of formal reduction potentials, neglecting in a first approach changes in activity coefficients due to ionic strength, the Nernst equation has to be applied taking care to first express the relationship as a function of pH. The second factor to be considered are the values of the concentrations taken into account in the Nernst equation. To define a formal reduction potential for a biochemical reaction, the pH value, the concentrations values and the hypotheses made on the activity coefficients must always be explicitly indicated. When using, or comparing, several formal reduction potentials they must also be internally consistent.
Problems may occur when mixing different sources of data using different conventions or approximations (i.e., with different underlying hypotheses). When working at the frontier between inorganic and biological processes (e.g., when comparing abiotic and biotic processes in geochemistry when microbial activity could also be at work in the system), care must be taken not to inadvertently directly mix standard reduction potentials versus SHE (pH = 0) with formal reduction potentials (pH = 7). Definitions must be clearly expressed and carefully controlled, especially if the sources of data are different and arise from different fields (e.g., picking and mixing data from classical electrochemistry and microbiology textbooks without paying attention to the different conventions on which they are based).
Examples with a Pourbaix diagram
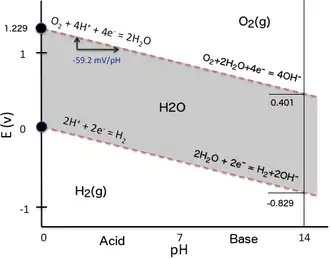
To illustrate the dependency of the reduction potential on pH, one can simply consider the two oxido-reduction equilibria determining the water stability domain in a Pourbaix diagram (Eh–pH plot). When water is submitted to electrolysis by applying a sufficient difference of electrical potential between two electrodes immersed in water, hydrogen is produced at the cathode (reduction of water protons) while oxygen is formed at the anode (oxidation of water oxygen atoms). The same may occur if a reductant stronger than hydrogen (e.g., metallic Na) or an oxidant stronger than oxygen (e.g., F2) enters in contact with water and reacts with it. In the Eh–pH plot here beside (the simplest possible version of a Pourbaix diagram), the water stability domain (grey surface) is delimited in term of redox potential by two inclined red dashed lines:
- Lower stability line with hydrogen gas evolution due to the proton reduction at very low Eh:
- 2 H+ + 2 e− ⇌ H2 (cathode: reduction)
- Higher stability line with oxygen gas evolution due to water oxygen oxidation at very high Eh:
- 2 H2O ⇌ O2 + 4 H+ + 4 e− (anode: oxidation)
When solving the Nernst equation for each corresponding reduction reaction (need to revert the water oxidation reaction producing oxygen), both equations have a similar form because the number of protons and the number of electrons involved within a reaction are the same and their ratio is one (2 H+/2 e− for H2 and 4 H+/4 e− with O2 respectively), so it simplifies when solving the Nernst equation expressed as a function of pH.
The result can be numerically expressed as follows:

Note that the slopes of the two water stability domain upper and lower lines are the same (-59.16 mV/pH unit), so they are parallel on a Pourbaix diagram. As the slopes are negative, at high pH, both hydrogen and oxygen evolution requires a much lower reduction potential than at low pH.
For the reduction of H+ into H2 the here above mentioned relationship becomes:
-
because by convention = 0 V for the standard hydrogen electrode (SHE: pH = 1).
So, at pH = 7, = -0.414 V for the reduction of protons.

For the reduction of O2 into 2 H2O the here above mentioned relationship becomes:
-
because = +1.229 V with respect to the standard hydrogen electrode (SHE: pH = 1).
So, at pH = 7, = +0.815 V for the reduction of oxygen.

The offset of -414 mV in is the same for both reduction reactions because they share the same linear relationship as a function of pH and the slopes of their lines are the same. This can be directly verified on a Pourbaix diagram. For other reduction reactions, the value of the formal reduction potential at a pH of 7, commonly referred for biochemical reactions, also depends on the slope of the corresponding line in a Pourbaix diagram i.e. on the ratio h⁄z of the number of H+ to the number of e− involved in the reduction reaction, and thus on the stoichiometry of the half-reaction. The determination of the formal reduction potential at pH = 7 for a given biochemical half-reaction requires thus to calculate it with the corresponding Nernst equation as a function of pH. One cannot simply apply an offset of -414 mV to the Eh value (SHE) when the ratio h⁄z differs from 1.
Applications in biology
Beside important redox reactions in biochemistry and microbiology, the Nernst equation is also used in physiology for calculating the electric potential of a cell membrane with respect to one type of ion. It can be linked to the acid dissociation constant.
Nernst potential
The Nernst equation has a physiological application when used to calculate the potential of an ion of charge z across a membrane. This potential is determined using the concentration of the ion both inside and outside the cell:
![{\displaystyle E={\frac {RT}{zF}}\ln {\frac {[{\text{ion outside cell}}]}{[{\text{ion inside cell}}]}}=2.3026{\frac {RT}{zF}}\log _{10}{\frac {[{\text{ion outside cell}}]}{[{\text{ion inside cell}}]}}.}](../I/18b6879b93f105e3024fdc947bf84fc7c70657d9.svg)
When the membrane is in thermodynamic equilibrium (i.e., no net flux of ions), and if the cell is permeable to only one ion, then the membrane potential must be equal to the Nernst potential for that ion.
Goldman equation
When the membrane is permeable to more than one ion, as is inevitably the case, the resting potential can be determined from the Goldman equation, which is a solution of G-H-K influx equation under the constraints that total current density driven by electrochemical force is zero:
![{\displaystyle E_{\mathrm {m} }={\frac {RT}{F}}\ln {\left({\frac {\displaystyle \sum _{i}^{N}P_{\mathrm {M} _{i}^{+}}\left[\mathrm {M} _{i}^{+}\right]_{\mathrm {out} }+\displaystyle \sum _{j}^{M}P_{\mathrm {A} _{j}^{-}}\left[\mathrm {A} _{j}^{-}\right]_{\mathrm {in} }}{\displaystyle \sum _{i}^{N}P_{\mathrm {M} _{i}^{+}}\left[\mathrm {M} _{i}^{+}\right]_{\mathrm {in} }+\displaystyle \sum _{j}^{M}P_{\mathrm {A} _{j}^{-}}\left[\mathrm {A} _{j}^{-}\right]_{\mathrm {out} }}}\right)},}](../I/391418bc185a0e25cad47d27335e2e6c9798e0da.svg)
where
- Em is the membrane potential (in volts, equivalent to joules per coulomb),
- Pion is the permeability for that ion (in meters per second),
- [ion]out is the extracellular concentration of that ion (in moles per cubic meter, to match the other SI units, though the units strictly don't matter, as the ion concentration terms become a dimensionless ratio),
- [ion]in is the intracellular concentration of that ion (in moles per cubic meter),
- R is the ideal gas constant (joules per kelvin per mole),
- T is the temperature in kelvins,
- F is the Faraday's constant (coulombs per mole).
The potential across the cell membrane that exactly opposes net diffusion of a particular ion through the membrane is called the Nernst potential for that ion. As seen above, the magnitude of the Nernst potential is determined by the ratio of the concentrations of that specific ion on the two sides of the membrane. The greater this ratio the greater the tendency for the ion to diffuse in one direction, and therefore the greater the Nernst potential required to prevent the diffusion. A similar expression exists that includes r (the absolute value of the transport ratio). This takes transporters with unequal exchanges into account. See: sodium-potassium pump where the transport ratio would be 2/3, so r equals 1.5 in the formula below. The reason why we insert a factor r = 1.5 here is that current density by electrochemical force Je.c.(Na+) + Je.c.(K+) is no longer zero, but rather Je.c.(Na+) + 1.5Je.c.(K+) = 0 (as for both ions flux by electrochemical force is compensated by that by the pump, i.e. Je.c. = −Jpump), altering the constraints for applying GHK equation. The other variables are the same as above. The following example includes two ions: potassium (K+) and sodium (Na+). Chloride is assumed to be in equilibrium.
![{\displaystyle E_{m}={\frac {RT}{F}}\ln {\left({\frac {rP_{\mathrm {K} ^{+}}\left[\mathrm {K} ^{+}\right]_{\mathrm {out} }+P_{\mathrm {Na} ^{+}}\left[\mathrm {Na} ^{+}\right]_{\mathrm {out} }}{rP_{\mathrm {K} ^{+}}\left[\mathrm {K} ^{+}\right]_{\mathrm {in} }+P_{\mathrm {Na} ^{+}}\left[\mathrm {Na} ^{+}\right]_{\mathrm {in} }}}\right)}.}](../I/c1878e58ef5d8d47cf178b68b4142a73e821691f.svg)
When chloride (Cl−) is taken into account,
![{\displaystyle E_{m}={\frac {RT}{F}}\ln {\left({\frac {rP_{\mathrm {K} ^{+}}\left[\mathrm {K} ^{+}\right]_{\mathrm {out} }+P_{\mathrm {Na} ^{+}}\left[\mathrm {Na} ^{+}\right]_{\mathrm {out} }+P_{\mathrm {Cl} ^{-}}\left[\mathrm {Cl} ^{-}\right]_{\mathrm {in} }}{rP_{\mathrm {K} ^{+}}\left[\mathrm {K} ^{+}\right]_{\mathrm {in} }+P_{\mathrm {Na} ^{+}}\left[\mathrm {Na} ^{+}\right]_{\mathrm {in} }+P_{\mathrm {Cl} ^{-}}\left[\mathrm {Cl} ^{-}\right]_{\mathrm {out} }}}\right)}.}](../I/fcc97595477c156d996f61cc4f0376847c0fba7b.svg)
Derivation
Using Boltzmann factor
For simplicity, we will consider a solution of redox-active molecules that undergo a one-electron reversible reaction
- Ox + e− ⇌ Red
and that have a standard potential of zero, and in which the activities are well represented by the concentrations (i.e. unit activity coefficient). The chemical potential μc of this solution is the difference between the energy barriers for taking electrons from and for giving electrons to the working electrode that is setting the solution's electrochemical potential. The ratio of oxidized to reduced molecules, [Ox]/[Red], is equivalent to the probability of being oxidized (giving electrons) over the probability of being reduced (taking electrons), which we can write in terms of the Boltzmann factor for these processes:
![{\displaystyle {\begin{aligned}{\frac {[\mathrm {Red} ]}{[\mathrm {Ox} ]}}&={\frac {\exp \left(-[{\text{barrier for gaining an electron}}]/kT\right)}{\exp \left(-[{\text{barrier for losing an electron}}]/kT\right)}}\\[6px]&=\exp \left({\frac {\mu _{\mathrm {c} }}{kT}}\right).\end{aligned}}}](../I/29207801fca2f2bf574f55c6c77b912f54d5c867.svg)
Taking the natural logarithm of both sides gives
![{\displaystyle \mu _{\mathrm {c} }=kT\ln {\frac {[\mathrm {Red} ]}{[\mathrm {Ox} ]}}.}](../I/32b863ec3018fd413c18c4af7cd43e4ac7374ab3.svg)
If μc ≠ 0 at [Ox]/[Red] = 1, we need to add in this additional constant:
![{\displaystyle \mu _{\mathrm {c} }=\mu _{\mathrm {c} }^{\ominus }+kT\ln {\frac {[\mathrm {Red} ]}{[\mathrm {Ox} ]}}.}](../I/a6b72c46af715358bfe31cebe3595215d119f3d6.svg)
Dividing the equation by e to convert from chemical potentials to electrode potentials, and remembering that k/e = R/F,[11] we obtain the Nernst equation for the one-electron process Ox + e− ⇌ Red :
![{\displaystyle {\begin{aligned}E&=E^{\ominus }-{\frac {kT}{e}}\ln {\frac {[\mathrm {Red} ]}{[\mathrm {Ox} ]}}\\&=E^{\ominus }-{\frac {RT}{F}}\ln {\frac {[\mathrm {Red} ]}{[\mathrm {Ox} ]}}.\end{aligned}}}](../I/332ed3cdb7d21fa792a53ac60015f5b3416ffc5a.svg)
Using thermodynamics (chemical potential)
Quantities here are given per molecule, not per mole, and so Boltzmann constant k and the electron charge e are used instead of the gas constant R and Faraday's constant F. To convert to the molar quantities given in most chemistry textbooks, it is simply necessary to multiply by the Avogadro constant: R = kNA and F = eNA. The entropy of a molecule is defined as

where Ω is the number of states available to the molecule. The number of states must vary linearly with the volume V of the system (here an idealized system is considered for better understanding, so that activities are posited very close to the true concentrations. Fundamental statistical proof of the mentioned linearity goes beyond the scope of this section, but to see this is true it is simpler to consider usual isothermal process for an ideal gas where the change of entropy ΔS = nR ln(V2/V1) takes place. It follows from the definition of entropy and from the condition of constant temperature and quantity of gas n that the change in the number of states must be proportional to the relative change in volume V2/V1. In this sense there is no difference in statistical properties of ideal gas atoms compared with the dissolved species of a solution with activity coefficients equaling one: particles freely "hang around" filling the provided volume), which is inversely proportional to the concentration c, so we can also write the entropy as

The change in entropy from some state 1 to another state 2 is therefore

so that the entropy of state 2 is

If state 1 is at standard conditions, in which c1 is unity (e.g., 1 atm or 1 M), it will merely cancel the units of c2. We can, therefore, write the entropy of an arbitrary molecule A as
![{\displaystyle S(\mathrm {A} )=S^{\ominus }(\mathrm {A} )-k\ln[\mathrm {A} ],}](../I/73acfd25accad61fa538780c535ef36c1d1e6c79.svg)
where is the entropy at standard conditions and [A] denotes the concentration of A. The change in entropy for a reaction

is then given by
![{\displaystyle \Delta S_{\mathrm {rxn} }={\big (}yS(\mathrm {Y} )+zS(\mathrm {Z} ){\big )}-{\big (}aS(\mathrm {A} )+bS(\mathrm {B} ){\big )}=\Delta S_{\mathrm {rxn} }^{\ominus }-k\ln {\frac {[\mathrm {Y} ]^{y}[\mathrm {Z} ]^{z}}{[\mathrm {A} ]^{a}[\mathrm {B} ]^{b}}}.}](../I/c35deda7d68ccb2c521c9e98bad604c9cec5b72a.svg)
We define the ratio in the last term as the reaction quotient:
![{\displaystyle Q_{r}={\frac {\displaystyle \prod _{j}a_{j}^{\nu _{j}}}{\displaystyle \prod _{i}a_{i}^{\nu _{i}}}}\approx {\frac {[\mathrm {Z} ]^{z}[\mathrm {Y} ]^{y}}{[\mathrm {A} ]^{a}[\mathrm {B} ]^{b}}},}](../I/271a9e2c5a6ed80d3d5cca079f9a794920a54897.svg)
where the numerator is a product of reaction product activities, aj, each raised to the power of a stoichiometric coefficient, νj, and the denominator is a similar product of reactant activities. All activities refer to a time t. Under certain circumstances (see chemical equilibrium) each activity term such as aνj
j may be replaced by a concentration term, [A].In an electrochemical cell, the cell potential E is the chemical potential available from redox reactions (E = μc/e). E is related to the Gibbs free energy change ΔG only by a constant:
ΔG = −zFE, where n is the number of electrons transferred and F is the Faraday constant. There is a negative sign because a spontaneous reaction has a negative Gibbs free energy ΔG and a positive potential E. The Gibbs free energy is related to the entropy by G = H − TS, where H is the enthalpy and T is the temperature of the system. Using these relations, we can now write the change in Gibbs free energy,

and the cell potential,

This is the more general form of the Nernst equation.
For the redox reaction Ox + z e− → Red,
![{\displaystyle Q_{r}={\frac {[\mathrm {Red} ]}{[\mathrm {Ox} ]}},}](../I/59fb8dd0b3884bbb390b4cbfacdc9cb81b37bca3.svg)
and we have:
![{\displaystyle {\begin{aligned}E&=E^{\ominus }-{\frac {kT}{ze}}\ln {\frac {[\mathrm {Red} ]}{[\mathrm {Ox} ]}}\\&=E^{\ominus }-{\frac {RT}{zF}}\ln {\frac {[\mathrm {Red} ]}{[\mathrm {Ox} ]}}\\&=E^{\ominus }-{\frac {RT}{zF}}\ln Q_{r}.\end{aligned}}}](../I/00d40bae8c6979f8de112935b38168b66a6ea932.svg)
The cell potential at standard temperature and pressure (STP) is often replaced by the formal potential , which includes the activity coefficients of the dissolved species under given experimental conditions (T, P, ionic strength, pH, and complexing agents) and is the potential that is actually measured in an electrochemical cell.


Relation to the chemical equilibrium
The standard Gibbs free energy is related to the equilibrium constant K as follows:[12]

At the same time, is also equal to the product of the total charge (zF) transferred during the reaction and the cell potential ():


The sign is negative, because the considered system performs the work and thus releases energy.
So,

And therefore:

Starting from the Nernst equation, one can also demonstrate the same relationship in the reverse way.
At chemical equilibrium, or thermodynamic equilibrium, the electrochemical potential (E) = 0 and therefore the reaction quotient (Qr) attains the special value known as the equilibrium constant (Keq):
- Qr = Keq
Therefore,

Or at standard state,

We have thus related the standard electrode potential and the equilibrium constant of a redox reaction.
Limitations
In dilute solutions, the Nernst equation can be expressed directly in the terms of concentrations (since activity coefficients are close to unity). But at higher concentrations, the true activities of the ions must be used. This complicates the use of the Nernst equation, since estimation of non-ideal activities of ions generally requires experimental measurements. The Nernst equation also only applies when there is no net current flow through the electrode. The activity of ions at the electrode surface changes when there is current flow, and there are additional overpotential and resistive loss terms which contribute to the measured potential.
At very low concentrations of the potential-determining ions, the potential predicted by Nernst equation approaches toward ±∞. This is physically meaningless because, under such conditions, the exchange current density becomes very low, and there may be no thermodynamic equilibrium necessary for Nernst equation to hold. The electrode is called unpoised in such case. Other effects tend to take control of the electrochemical behavior of the system, like the involvement of the solvated electron in electricity transfer and electrode equilibria, as analyzed by Alexander Frumkin and B. Damaskin,[13] Sergio Trasatti, etc.
Significance in other scientific fields
The Nernst equation has been involved in the scientific controversy about cold fusion. Fleischmann and Pons, claiming that cold fusion could exist, calculated that a palladium cathode immersed in a heavy water electrolysis cell could achieve up to 1027 atmospheres of pressure inside the crystal lattice of the metal of the cathode, enough pressure to cause spontaneous nuclear fusion. In reality, only 10,000–20,000 atmospheres were achieved. The American physicist John R. Huizenga claimed their original calculation was affected by a misinterpretation of the Nernst equation.[18] He cited a paper about Pd–Zr alloys.[19]
The Nernst equation allows the calculation of the extent of reaction between two redox systems and can be used, for example, to assess whether a particular reaction will go to completion or not. At chemical equilibrium, the electromotive forces (emf) of the two half cells are equal. This allows the equilibrium constant K of the reaction to be calculated and hence the extent of the reaction.
See also
- Concentration cell
- Dependency of reduction potential on pH
- Electrode potential
- Galvanic cell
- Goldman equation
- Membrane potential
- Nernst–Planck equation
- Pourbaix diagram
- Reduction potential
- Solvated electron
- Standard electrode potential
- Standard electrode potential (data page)
- Standard apparent reduction potentials in biochemistry at pH 7 (data page)
References
- Orna, Mary Virginia; Stock, John (1989). Electrochemistry, past and present. Columbus, OH: American Chemical Society. ISBN 978-0-8412-1572-6. OCLC 19124885.
- Wahl (2005). "A Short History of Electrochemistry". Galvanotechtnik. 96 (8): 1820–1828.
- Bard, Allen J.; Faulkner, Larry R. (2001). "Chapter 2. Potentials and Thermodynamics of Cells – See: 2.1.6 Formal Potentials". Electrochemical methods: Fundamentals and applications (2 ed.). New York: John Wiley & Sons. p. 52.
- Wenzel, Thomas (2020-06-09). "4. Table of Standard State Electrochemical Potentials". Chemistry LibreTexts. Retrieved 2021-11-24.
- Kano, Kenji (2002). "Redox potentials of proteins and other compounds of bioelectrochemical interest in aqueous solutions". Review of Polarography. 48 (1): 29–46. doi:10.5189/revpolarography.48.29. eISSN 1884-7692. ISSN 0034-6691. Retrieved 2021-12-02.
- "Formal potential". TheFreeDictionary.com. Retrieved 2021-12-06.
- PalmSens (2021). "Origins of electrochemical potentials — PalmSens". PalmSens. Retrieved 2021-12-06.
- Brown, Raymond A.; Swift, Ernest H. (1949). "The formal potential of the antimonous-antimonic half cell in hydrochloric acid solutions". Journal of the American Chemical Society. 71 (8): 2719–2723. ISSN 0002-7863.
Quote: A formal potential is defined as the potential of a half-cell, measured against the standard hydrogen electrode, when the total concentration of each oxidation state is one formal.
- Harvey, David (2020-06-15). "2.2: Concentration". Chemistry LibreTexts. Retrieved 2021-12-15.
- Voet, Donald; Voet, Judith G.; Pratt, Charlotte W. (2016). "Table 14-4 Standard Reduction Potentials for Some Biochemically Import Half-Reactions". Fundamentals of Biochemistry: Life at the Molecular Level (5th ed.). Wiley. p. 466. ISBN 978-1-118-91840-1.
- R = NAk; see gas constant
F = NAe; see Faraday constant - "20.5: Gibbs energy and redox reactions". Chemistry LibreTexts. 2014-11-18. Retrieved 2021-12-06.
- J. Electroanal. Chem., 79 (1977), 259-266
- Karaoglanoff, Z. (January 1906), "Über Oxydations- und Reduktionsvorgänge bei der Elektrolyse von Eisensaltzlösungen" [On Oxidation and Reduction Processes in the Electrolysis of Iron Salt Solutions], Zeitschrift für Elektrochemie (in German), 12 (1): 5–16, doi:10.1002/bbpc.19060120105
- Bard, Allen J.; Inzelt, György; Scholz, Fritz, eds. (2012-10-02), "Karaoglanoff equation", Electrochemical Dictionary, Springer, pp. 527–528, ISBN 9783642295515
- Zutshi, Kamala (2008), Introduction to Polarography and Allied Techniques, pp. 127–128, ISBN 9788122417913
- The Journal of Physical Chemistry, Volume 10, p 316. https://books.google.com/books?id=zCMSAAAAIAAJ&pg=PA316
- Huizenga, John R. (1993). Cold Fusion: The Scientific Fiasco of the Century (2 ed.). Oxford and New York: Oxford University Press. pp. 33, 47. ISBN 978-0-19-855817-0.
- Huot, J. Y. (1989). "Electrolytic Hydrogenation and Amorphization of Pd-Zr Alloys". Journal of the Electrochemical Society. 136 (3): 630–635. doi:10.1149/1.2096700. ISSN 0013-4651.
External links
- Nernst/Goldman Equation Simulator
- Nernst Equation Calculator
- Interactive Nernst/Goldman Java Applet
- DoITPoMS Teaching and Learning Package- "The Nernst Equation and Pourbaix Diagrams"
- "20.5: Gibbs energy and redox reactions". Chemistry LibreTexts. 2014-11-18. Retrieved 2021-12-06.