MID1
MID1 is a protein that belongs to the Tripartite motif family (TRIM) and is also known as TRIM18.[5][6] The MID1 gene is located on the short arm of the X chromosome and loss-of-function mutations in this gene are causative of the X-linked form of a rare developmental disease, Opitz G/BBB Syndrome.[5][7]
| MID1 | |||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||||||||
| Aliases | MID1, BBBG1, FXY, GBBB1, MIDIN, OGS1, OS, OSX, RNF59, TRIM18, XPRF, ZNFXY, midline 1, GBBB | ||||||||||||||||||||||||||||||
| External IDs | OMIM: 300552 MGI: 1100537 HomoloGene: 7837 GeneCards: MID1 | ||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||

The MID1 gene and its product
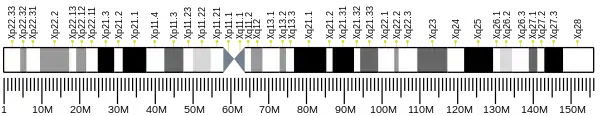
The human MID1 gene is located on the short arm of the X chromosome (Xp22.2) and includes 9 coding exons, spanning approximately 400 kb of the genome.[5][8] Upstream to the first coding exon, the MID1 gene employs alternative 5’ untranslated exons and at least five alternative promoters that drive the transcription of the gene, resulting in several MID1 transcript isoforms.[9] The MID1 gene encodes a 667 amino acid protein that belongs to the TRIM family. MID1 protein consists of a conserved N-terminal tripartite module composed of a RING domain, 2 B-Box domains (B-box 1 and B-box 2) and a coiled-coil region.[5][6] Within the TRIM family, MID1 belongs to the C-I subgroup characterised by the presence, downstream to the tripartite motif, of a COS domain, a Fibronectin type III (FN3) repeat and a PRY-SPRY domain.[10]
MID1 main cellular functions
MID1 as an E3 ubiquitin ligase
MID1 is a microtubular protein[11][12] that acts as an ubiquitin E3 ligase in vitro and in cells. Ubiquitination is a type of post-translational modification in which the transfer of one or several ubiquitin peptide molecules to substrates determines their stability and/or activity.[13] The MID1 E3 ubiquitin ligase activity is catalysed by the RING domain, a hallmark of one of the main classes of E3 ubiquitin ligases that, within the ubiquitination cascade, facilitate the transfer of the ubiquitin peptide to specific substrates.[14][15][16] Several MID1 E3 ubiquitin ligase targets have been reported: Alpha4 (α4) and its associated phosphatase, PP2A,[14] Fu,[17] Pax6[18] and BRAF35.[19]
MID1-α4-PP2A complex
Together with α4 and PP2A, MID1 can form a ternary complex in which α4 acts as an adaptor protein.[14] The data so far indicate that MID1 promotes α4 mono-ubiquitination, leading to its calpain-dependent cleavage[20] that in turn causes PP2A catalytic subunit (PP2Ac) polyubiquitination and proteasomal degradation.[14] Since PP2A is involved in many cellular processes,[21] the MID1-α4-PP2A ternary complex may be involved in the regulation of several of them, mainly on microtubules. The complex can modulate mTORC1 signalling; indeed PP2A attenuates mTORC1 activity through dephosphorylation. By lowering PP2Ac levels, MID1 leads to increase mTORC1 signalling.[22] Conversely, the lack or loss-of-function mutations of MID1 lead to increased levels of PP2A and, as a consequence, to a general hypo-phosphorylation of PP2A targets, included mTORC1. The signalling of mTORC1 is implicated in cytoskeletal dynamics, intracellular transport, cell migration, autophagy, protein synthesis, cell metabolism, so it is possible that MID1, by controlling PP2Ac, is ultimately implicated in some of these cellular processes.
MID1 and Sonic Hedgehog
MID1 is also involved is the Sonic Hedgehog (Shh) pathway.[23] MID1 catalyses the ubiquitination and proteasomal-dependent cleavage of Fu, a kinase involved in Hedgehog signalling pathway.[17] The cleavage of the kinase domain of Fu favours the translocation of the transcription factor GLI3A (activator form) in the nucleus.[17][24] In this way, GLI3A activates the expression of Shh target genes, leading to an increase of Shh signalling. The cross talk between MID1 and the Shh pathway is also supported by experimental evidence in model organisms.[18][25]
Role and expression during embryonic development
MID1 is nearly ubiquitously expressed in all embryonic tissues, having an important function during development. Several model organisms have been used to study the expression pattern of MID1 transcript at different times of gestation: mouse,[26][27] chicken,[28][29] xenopus[18][30] and also human embryos.[31] At the very early stage of embryonic development, MID1 is expressed in the primitive node where MID1 plays a pivotal role in establishing the molecular asymmetry at the node, which is crucial for the early definition of the laterality as embryonic development progresses. Later in embryogenesis, at the neurulation stage, MID1 transcript is mainly observed in the cranial region of the developing neural folds. Starting from midgestation, the highest levels of MID1 transcript are observed in the proliferating compartments of the central nervous system and in the epithelia of the developing branchial arches, craniofacial processes, optic vesicle, in the heart and in the gastrointestinal and urogenital system.
Clinical significance
The MID1 gene was identified concomitantly with the discovery that it was causatively mutated in patients with a rare genetic disease, the X-linked form of Opitz G/BBB syndrome (XLOS) (OMIM #300000).[5] XLOS is a congenital malformative disorder characterised by defects in the embryonic development of midline structures. XLOS is characterised by high variability of the clinical signs and, being X-linked, males are generally affected. The most frequently observed signs are: dysmorphic features, mainly represented by hypertelorism often associated with cleft lip and palate, frontal bossing, large nasal bridge, and low-set ears. Laryngo-tracheo-esophageal abnormalities are also frequently observed in XLOS patients as well as external genitalia abnormalities that are predominantly represented by various-degree-hypospadias.[32] In addition, XLOS patients can present cardiac abnormalities and anal defects.[32] XLOS also shows a neurological component represented by cerebellar vermis hypoplasia and agenesis or hypoplasia of the corpus callosum accompanied by intellectual disabilities and developmental delays.[32] Since its discovery as the causative gene for XLOS, approximately one hundred different pathogenetic mutations have been described in the MID1 gene. Even though the type and the distribution of mutations suggested a loss-of-function mechanism in the pathogenesis of Opitz syndrome, the aetiology of the disease remains still unclear. Additional clinical conditions are described to be associated with alterations of MID1, given also its implication in a wide variety of cellular mechanisms. In fact, involvement of MID1 in asthma, cancer, and neurodegeneration relevant pathways has been reported.
Notes
References
- GRCh38: Ensembl release 89: ENSG00000101871 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000035299 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, et al. (November 1997). "Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22". Nature Genetics. 17 (3): 285–91. doi:10.1038/ng1197-285. hdl:2066/24575. PMID 9354791. S2CID 5832037.
- Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. (May 2001). "The tripartite motif family identifies cell compartments". The EMBO Journal. 20 (9): 2140–51. doi:10.1093/emboj/20.9.2140. PMC 125245. PMID 11331580.
- Opitz JM (October 1987). "G syndrome (hypertelorism with esophageal abnormality and hypospadias, or hypospadias-dysphagia, or "Opitz-Frias" or "Opitz-G" syndrome)--perspective in 1987 and bibliography". American Journal of Medical Genetics. 28 (2): 275–85. doi:10.1002/ajmg.1320280203. PMID 3322001.
- Van den Veyver IB, Cormier TA, Jurecic V, Baldini A, Zoghbi HY (July 1998). "Characterization and physical mapping in human and mouse of a novel RING finger gene in Xp22". Genomics. 51 (2): 251–61. doi:10.1006/geno.1998.5350. PMID 9722948.
- Landry JR, Mager DL (November 2002). "Widely spaced alternative promoters, conserved between human and rodent, control expression of the Opitz syndrome gene MID1". Genomics. 80 (5): 499–508. doi:10.1006/geno.2002.6863. PMID 12408967.
- Short KM, Cox TC (March 2006). "Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding". The Journal of Biological Chemistry. 281 (13): 8970–80. doi:10.1074/jbc.M512755200. PMID 16434393.
- Schweiger S, Foerster J, Lehmann T, Suckow V, Muller YA, Walter G, et al. (March 1999). "The Opitz syndrome gene product, MID1, associates with microtubules". Proceedings of the National Academy of Sciences of the United States of America. 96 (6): 2794–9. Bibcode:1999PNAS...96.2794S. doi:10.1073/pnas.96.6.2794. PMC 15848. PMID 10077590.
- Cainarca S, Messali S, Ballabio A, Meroni G (August 1999). "Functional characterization of the Opitz syndrome gene product (midin): evidence for homodimerization and association with microtubules throughout the cell cycle". Human Molecular Genetics. 8 (8): 1387–96. doi:10.1093/hmg/8.8.1387. PMID 10400985.
- Hershko A, Ciechanover A (1998). "The ubiquitin system". Annual Review of Biochemistry. 67: 425–79. doi:10.1146/annurev.biochem.67.1.425. PMID 9759494.
- Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S, Ropers HH, et al. (November 2001). "MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation". Nature Genetics. 29 (3): 287–94. doi:10.1038/ng762. PMID 11685209. S2CID 34834612.
- Han X, Du H, Massiah MA (April 2011). "Detection and characterization of the in vitro e3 ligase activity of the human MID1 protein". Journal of Molecular Biology. 407 (4): 505–20. doi:10.1016/j.jmb.2011.01.048. PMID 21296087.
- Napolitano LM, Jaffray EG, Hay RT, Meroni G (March 2011). "Functional interactions between ubiquitin E2 enzymes and TRIM proteins" (PDF). The Biochemical Journal. 434 (2): 309–19. doi:10.1042/BJ20101487. PMID 21143188. S2CID 5932399.
- Schweiger S, Dorn S, Fuchs M, Köhler A, Matthes F, Müller EC, et al. (November 2014). "The E3 ubiquitin ligase MID1 catalyzes ubiquitination and cleavage of Fu". The Journal of Biological Chemistry. 289 (46): 31805–17. doi:10.1074/jbc.M113.541219. PMC 4231658. PMID 25278022.
- Pfirrmann T, Jandt E, Ranft S, Lokapally A, Neuhaus H, Perron M, Hollemann T (September 2016). "Hedgehog-dependent E3-ligase Midline1 regulates ubiquitin-mediated proteasomal degradation of Pax6 during visual system development". Proceedings of the National Academy of Sciences of the United States of America. 113 (36): 10103–8. Bibcode:2016PNAS..11310103P. doi:10.1073/pnas.1600770113. PMC 5018744. PMID 27555585.
- Zanchetta ME, Napolitano LM, Maddalo D, Meroni G (October 2017). "The E3 ubiquitin ligase MID1/TRIM18 promotes atypical ubiquitination of the BRCA2-associated factor 35, BRAF35". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1864 (10): 1844–1854. doi:10.1016/j.bbamcr.2017.07.014. PMID 28760657.
- Watkins GR, Wang N, Mazalouskas MD, Gomez RJ, Guthrie CR, Kraemer BC, et al. (July 2012). "Monoubiquitination promotes calpain cleavage of the protein phosphatase 2A (PP2A) regulatory subunit α4, altering PP2A stability and microtubule-associated protein phosphorylation". The Journal of Biological Chemistry. 287 (29): 24207–15. doi:10.1074/jbc.M112.368613. PMC 3397847. PMID 22613722.
- Sontag E (January 2001). "Protein phosphatase 2A: the Trojan Horse of cellular signaling". Cellular Signalling. 13 (1): 7–16. doi:10.1016/s0898-6568(00)00123-6. PMID 11257442.
- Liu E, Knutzen CA, Krauss S, Schweiger S, Chiang GG (May 2011). "Control of mTORC1 signaling by the Opitz syndrome protein MID1". Proceedings of the National Academy of Sciences of the United States of America. 108 (21): 8680–5. Bibcode:2011PNAS..108.8680L. doi:10.1073/pnas.1100131108. PMC 3102420. PMID 21555591.
- Ingham PW, McMahon AP (December 2001). "Hedgehog signaling in animal development: paradigms and principles". Genes & Development. 15 (23): 3059–87. doi:10.1101/gad.938601. PMID 11731473.
- Krauss S, Foerster J, Schneider R, Schweiger S (June 2008). "Protein phosphatase 2A and rapamycin regulate the nuclear localization and activity of the transcription factor GLI3". Cancer Research. 68 (12): 4658–65. doi:10.1158/0008-5472.CAN-07-6174. PMID 18559511.
- Granata A, Quaderi NA (June 2003). "The Opitz syndrome gene MID1 is essential for establishing asymmetric gene expression in Hensen's node". Developmental Biology. 258 (2): 397–405. doi:10.1016/s0012-1606(03)00131-3. PMID 12798296.
- Dal Zotto L, Quaderi NA, Elliott R, Lingerfelter PA, Carrel L, Valsecchi V, et al. (March 1998). "The mouse Mid1 gene: implications for the pathogenesis of Opitz syndrome and the evolution of the mammalian pseudoautosomal region". Human Molecular Genetics. 7 (3): 489–99. doi:10.1093/hmg/7.3.489. PMID 9467009.
- Lancioni A, Pizzo M, Fontanella B, Ferrentino R, Napolitano LM, De Leonibus E, Meroni G (February 2010). "Lack of Mid1, the mouse ortholog of the Opitz syndrome gene, causes abnormal development of the anterior cerebellar vermis". The Journal of Neuroscience. 30 (8): 2880–7. doi:10.1523/JNEUROSCI.4196-09.2010. PMC 6633954. PMID 20181585.
- Richman JM, Fu KK, Cox LL, Sibbons JP, Cox TC (2002). "Isolation and characterisation of the chick orthologue of the Opitz syndrome gene, Mid1, supports a conserved role in vertebrate development". The International Journal of Developmental Biology. 46 (4): 441–8. PMID 12141430.
- Latta EJ, Golding JP (2011). "Regulation of PP2A activity by Mid1 controls cranial neural crest speed and gangliogenesis". Mechanisms of Development. 128 (11–12): 560–76. doi:10.1016/j.mod.2012.01.002. PMID 22285438.
- Suzuki M, Hara Y, Takagi C, Yamamoto TS, Ueno N (July 2010). "MID1 and MID2 are required for Xenopus neural tube closure through the regulation of microtubule organization". Development. 137 (14): 2329–39. doi:10.1242/dev.048769. PMID 20534674.
- Pinson L, Augé J, Audollent S, Mattéi G, Etchevers H, Gigarel N, et al. (May 2004). "Embryonic expression of the human MID1 gene and its mutations in Opitz syndrome". Journal of Medical Genetics. 41 (5): 381–6. doi:10.1136/jmg.2003.014829. PMC 1735763. PMID 15121778.
- Fontanella B, Russolillo G, Meroni G (May 2008). "MID1 mutations in patients with X-linked Opitz G/BBB syndrome". Human Mutation. 29 (5): 584–94. doi:10.1002/humu.20706. PMID 18360914.
Further reading
- Meroni, Germana (1993). "X-Linked Opitz G/BBB Syndrome". GeneReviews®. University of Washington, Seattle. PMID 20301502.
PDB gallery | |
|---|---|
|