Isolated hyperchlorhidrosis
Isolated hyperchlorhidrosis, also known as Carbonic anhydrase XII deficiency, is a rare autosomal recessive genetic condition characterized by a lifelong tendency to lose massive amounts of sodium and chloride through sweat which leads to various symptoms.[1]
| Isolated hyperchlorhidrosis | |
|---|---|
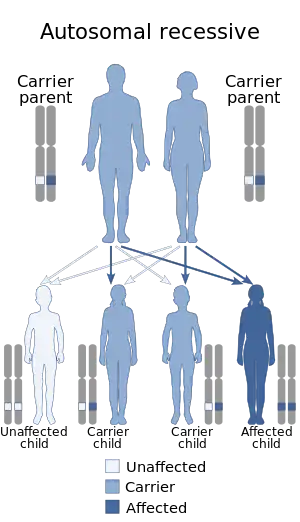 | |
| This condition is inherited in an autosomal recessive manner. | |
| Specialty | Medical genetics |
| Symptoms | Loss of salt through sweat |
| Complications | Hypokalemic dehydration |
| Usual onset | Infancy |
| Duration | Lifelong |
Presentation
Symptoms from the loss of salt typically start presenting during infancy, and they include episodic hyponatremic dehydration and bouts of vomiting and/or diarrhea which typically occur after these episodes. Other symptoms include failure to thrive which usually resolves by early childhood.[2]
Laboratory tests done on patients with this condition typically show hyperkalemia, high levels of aldosterone, and high levels of chloride and sodium found in a patient's sweat.[3]
Complications
Warm environments and physical exercise may trigger an episode of hyponatremia due to the sweating that is typically associated with said scenarios.[2]
Genetics
This condition is associated with the CA12 gene, located in the 15th chromosome. This gene plays a role in the production of a protein known as carbonic anhydrase 12, the family of proteins (carbonic anhydrase) this protein belongs to is responsible for the production of bicarbonate ions and protons which affect the levels of pH in the cells of the body, which by themselves might regulate the amount of sodium that leaves the body with sweat.[4]
The mutation involved in this condition is known as E143K or Glu143Lys, and it involves an amino acid substitution from glutamate to lysine at position 143 of the CA12 protein. This change results in a loss-of-function mutation, and it impairs more than half (70%) of the function this protein naturally possesses, this reduces the protein's ability of regulating pH levels, which, as previously mentioned, are responsible for regulating how much salt leaves the body during sweating, resulting in the loss of salt through sweat characteristic of this condition.[4]
Diagnosis
This condition is usually diagnosed through laboratory tests and genetic testing.[5]
Other conditions that can cause loss of salt in the sweat – such as cystic fibrosis or pseudohypoaldosteronism, type IB1, have to be excluded, since this condition, like the name implies, occurs by itself and in a manner that is isolated and unrelated to other conditions.[2]
Treatment
Provision of sodium chloride for affected people was proven to be successful at treating symptoms associated to this condition with the Bedouin patients reported by Feldshtein et al. (2010)[6] and Muhammad et al. (2011).[7]
Epidemiology
Around 13 cases from 7 families worldwide have been described in medical literature.[5]
The condition is the most common among the Bedouin people of Israel, particularly among families that practice familial endogamy.[2][5]
History
The following list include cases of isolated hyperchlorhidrosis that have been described in medical literature
1979
The condition was first discovered in 1979, when Greenburg et al. described 2 Puerto Rican brothers ages 7 and 15, respectively, who were found to have high amounts of chloride in their sweat. No disorder (including cystic fibrosis) that could be responsible for this symptom was identified in neither of the brothers, although one of them were found to have Klinefelter syndrome upon karyotyping.[8]
2010
In 2010, Feldshtein et al. described 7 affected members belonging to a multigenerational Israeli Bedouin family. Alongside the hyperchlorhidrosis, said patients were found to have gastroenteritis. Personal testimonies from the patients told that high amounts of visible salt deposits could be spotted during sweating, particularly on areas of the body like the nape. A genome-wide linkage study done on the affected members mapped this condition's locus to a 11-cM region in chromosome 15. The disease-related haplotype was homozygous for those members of the family that were affected and either heterozygous or not present for unaffected members of the family. Sequencing of candidate genes within this genetic region found a homozygous missense mutation in exon 4 of the CA12 gene in affected members (heterozygous in some non-affected members).[6]
2011
In 2011, Muhammad et al. described 3 patients from 3 consanguineous Bedouin families related through the same small clan with failure to thrive and, in 2 of the 3 patients, gastroenteritis. Laboratory tests found the associated bodily abnormalities of the disorder (hyponatremia, hyperkalemia, increased aldosterone, and increased sweat chloride, to be exact). Treatment with oral supplementation of sodium chloride proved successful, although the patients who accepted the treatment (2 out of the 3) developed benign salt cravings at ages 5.5 and 6. The patient who declined treatment continued having episodes of hyponatremic dehydration. All three of the patients showed "catch-up growth" as compensation for their previous failure at thriving. Sequencing of candidate genes following genome-wide linkage analysis found the exact same homozygous, missense mutation in the CA12 gene previously described in medical literature.[7]
2016
In 2016, Lee et al. describes 3 patients from 2 un-related families of Euro-American and Omani descent, respectively.[9]
The first patient is a 25-year-old white woman from the United States who had the characteristic failure to thrive and episodic hyponatremia associated with this condition. In addition to these symptoms, she also had axillary hyperhidrosis (which was treated by a dermatologist) and symptoms suggestive of cystic fibrosis. To be more specific, she had childhood-onset airway obstruction and chronic cough associated with pseudomonas infections. She also had mild bronchiectasis and airway dilatation, both findings of which were found through high-resolution CT imaging. Testing of her CFTR (gene associated with cystic fibrosis), SCNN1A, SCNN1B, and SCNN1G genes came back normal. Sequencing of her CA12 gene found two pathogenic variants: a paternally-inherited c.908-1 G>A substitution and a maternally-inherited four-nucleotide insertion (c.859_860insACCT). On the Exome Aggregation Consortium variant browser, only 53 people were found to carry her c.908-1 G>A substitution mutation, while none were found to carry her four-nucleotide insertion. Saguer sequencing of the gene on the family members of the woman found the same mutations on their respective family sides.[9]
The second and third patients reported by Lee et al. were two siblings, born to consanguineous Omani parents. In addition to the two most common findings of isolated hyperchlorhidrosis, they were also found to have hyperkeratosis affecting their heels. Their lungs and their respective function were found to be healthy. Sequencing of the patients' CA12 gene found a c.363 C>A missense mutation in exon 4 of the gene that changed the His at codon 124 of the gene to Gln. Sequencing of the CA12 gene of the patients' three other unaffected siblings and two unaffected parents found the mutation in its heterozygous state on one of their siblings and both of their parents, while only two of their siblings carried both wild-type (healthy) variants of the gene.[9]
References
- "Isolated hyperchlorhidrosis – NIH Genetic Testing Registry (GTR) – NCBI". www.ncbi.nlm.nih.gov. Retrieved 26 December 2022.
- "Isolated hyperchlorhidrosis: MedlinePlus Genetics". medlineplus.gov. Retrieved 26 December 2022.
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Isolated hyperchlorhidrosis". www.orpha.net. Retrieved 26 December 2022.
- "CA12 gene: MedlinePlus Genetics". medlineplus.gov. Retrieved 26 December 2022.
- "Entry – #143860 – HYPERCHLORHIDROSIS, ISOLATED; HYCHL – OMIM". www.omim.org. Retrieved 26 December 2022.
- Feldshtein, Maya; Elkrinawi, Suliman; Yerushalmi, Baruch; Marcus, Barak; Vullo, Daniela; Romi, Hila; Ofir, Rivka; Landau, Daniel; Sivan, Sara; Supuran, Claudiu T.; Birk, Ohad S. (12 November 2010). "Hyperchlorhidrosis caused by homozygous mutation in CA12, encoding carbonic anhydrase XII". American Journal of Human Genetics. 87 (5): 713–720. doi:10.1016/j.ajhg.2010.10.008. ISSN 1537-6605. PMC 2978943. PMID 21035102.
- Muhammad, Emad; Leventhal, Neta; Parvari, Galit; Hanukoglu, Aaron; Hanukoglu, Israel; Chalifa-Caspi, Vered; Feinstein, Yael; Weinbrand, Jenny; Jacoby, Harel; Manor, Esther; Nagar, Tal; Beck, John C.; Sheffield, Val C.; Hershkovitz, Eli; Parvari, Ruti (April 2011). "Autosomal recessive hyponatremia due to isolated salt wasting in sweat associated with a mutation in the active site of Carbonic Anhydrase 12". Human Genetics. 129 (4): 397–405. doi:10.1007/s00439-010-0930-4. ISSN 1432-1203. PMID 21184099.
- Greenburg, F., Schidlow, D., Palmer, N., Huang, N. Isolated hyperchlorhidrosis without evidence of cystic fibrosis in two brothers, a possible autosomal recessive trait. (Abstract) Am. J. Hum. Genet. 31: 73A only, 1979.
- Lee, Melissa; Vecchio-Pagán, Briana; Sharma, Neeraj; Waheed, Abdul; Li, Xiaopeng; Raraigh, Karen S.; Robbins, Sarah; Han, Sangwoo T.; Franca, Arianna L.; Pellicore, Matthew J.; Evans, Taylor A.; Arcara, Kristin M.; Nguyen, Hien; Luan, Shan; Belchis, Deborah (15 May 2016). "Loss of carbonic anhydrase XII function in individuals with elevated sweat chloride concentration and pulmonary airway disease". Human Molecular Genetics. 25 (10): 1923–1933. doi:10.1093/hmg/ddw065. ISSN 1460-2083. PMC 5062583. PMID 26911677.