Intravascular lymphomas
Intravascular lymphomas (IVL) are rare cancers in which malignant lymphocytes proliferate and accumulate within blood vessels. Almost all other types of lymphoma involve the proliferation and accumulation of malignant lymphocytes in lymph nodes, other parts of the lymphatic system (e.g. the spleen), and various non-lymphatic organs (e.g. bone marrow and liver) but not in blood vessels.[1]
| Intravascular lymphomas | |
|---|---|
| Other names | Subtypes: intravascular large B-cell lymphoma; intravascular NK/T-cell lymphoma; intravascular NK-cell lymphoma; Intravasular T-cell lymphoma |
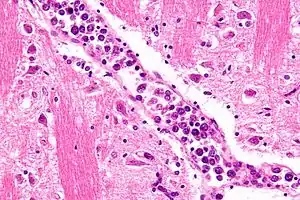 | |
| Micrograph showing an intravascular large B-cell lymphoma in a blood vessel of the brain. H&E stain. | |
| Specialty | Hematology, oncology, dermatology |
| Causes | Epstein–Barr virus for intravascular NK- and T-cell lymphomas |
| Prognosis | Guarded |
IVL fall into three different forms based on the type of lymphocyte causing the disease. Intravascular large B-cell lymphoma (IVBCL), which constitutes ~90% of all IVL, is a lymphoma of malignant B-cell lymphocytes[2] as classified by the World Health Organization, 2016.[3] The remaining IVL types, which have not yet been formally classified by the World Health Organization, are defined based mainly on case reports;[1] these IVL are 1) intravascular NK-cell lymphoma (IVNKL) in which the malignant cells are a type of T cell lymphocyte termed natural killer cells (NK-cells) and 2) intravascular T-cell lymphoma (IVTL) in which the neoplastic cells are primarily, if not exclusively, a type of t-cell termed cytotoxic T-cells. Because of their similarities and extreme rarities, IVL lymphomas caused by NK-cells and cytotoxic T-cells are often grouped together under the term intravascular NK/T cell lymphomas (IVNK/TL).[4] The malignant cells in IVNK/TL are typically infected with the Epstein–Barr virus suggesting that these lymphomas are examples of the Epstein-Barr virus-associated lymphoproliferative diseases.[4] Since infection with this virus is rarely seen in IVBCL, this form of IVL is not typically regarded as one of the Epstein-Barr virus-associated lymphoproliferative diseases.[2]
Intravascular large B-cell[5] and intravascular NK/T cell[4] IVL are typically very aggressive lymphomas that afflict middle-aged and elderly adults. At the time of diagnosis, they accumulate within small-sized and medium-sized but not large-sized blood vessels of the skin, central nervous system, and, less frequently. virtually any other organ system. Unlike most lymphomas, however, they generally do not accumulate or infiltrate lymph nodes. All of the IVL are frequently associated with systemic B symptoms such as fever and weight loss, as well as symptoms related to the other organs in which they accumulate in blood vessels, constrict blood flow, and thereby cause severe damage due to infarction, i.e. damage due to the loss of blood flow.[4][5]
Historically, most cases of the intravascular lymphomas responded very poorly to standard chemotherapy regimens that were used to treat other types of the B-cell lymphomas. With few exceptions, these intravascular lymphomas progressed very rapidly. More recently, however, the addition to these chemotherapy regimens of the immunotherapy agents, Rituximab, which acts to kill B-cells, has greatly improved their effectiveness and thereby the prognosis of the most common form of these diseases, the intravascular B-cell lymphomas.[5] Unfortunately, no such agent that is directed against NK-cells or cytotoxic T-cells has yet been reported to be useful in treating these two types of the intravascular B-cell lymphomas.
History
In 1959, Pfleger and Tappeiner first reported on a cancer in which malignant cells grew uncontrollably within the lumen of blood vessels; the authors suggested that these malignant cells were derived from the endothelial cells lining the vasculature and therefore termed the disorder angioendotheliomatosis proliferans systemisata.[6] Subsequent studies reported in 1982, 1985, and 1986 led to the conclusion that these malignant cells were derived from lymphocytes rather than endothelial cells. These along with other studies termed the disease angioendotheliomatosis, neoplastic angiotheliomatosis, intravascular lymphomatosis, angioendotheliotropic (intravascular) lymphoma, angiotropic large-cell lymphoma, diffuse large-cell lymphoma,[7] intralymphatic lymphomatosis, and, less specifically, malignant angioendotheliomatosis or intravascular lymphoma. By 2001, the World Health Organization had defined the disease as a malignant B-cell lymphoma termed intravascular large B-cell lymphoma.
Santucci et al. first reported a case of IVL that involved malignant NK cells. Some 2 dozen other cases of intravascular NK cell lymphoma have been reported by 2018.[8] In 2008, 29 case reports of purported intravascular T-cell lymphoma were reviewed; only two of these cases were associated with evidence strongly suggesting that the malignant cells were cytotoxic T-cells. Subsequently, a few more cases of cytotoxic T-cell-based have been reported. There remains a possibility that future studies will find other T-cell types may cause IVTCL.[9]
Intravascular large B-cell lymphoma
Intravascular large B-cell lymphomas fall into three distinct variants, all of which involve the intravascular accumulation of malignant B-cells and appear to have a similar pathophysiology. However, they differ in the distribution of their lesions, types of populations affected, prognoses, and treatments. These three variants are: 1) intravascular large B-cell lymphoma classical, 2) intravascular large B-cell lymphoma, cutaneous variant, and 3) intravascular large B-cell lymphoma, hemophagocytic syndrome-associated variant.[5] The following sections give the common pathophysiology of the three variants and then describes the lesions, populations affected, prognoses, and treatments of each variant in separate sections.
Pathophysiology of the intravascular B-cell lymphomas
The gene, chromosome, and gene expression abnormalities in IVBCL have not been fully evaluated. Studies to date indicate that the malignant cells in this disease have mutations in their MYD88 (44% of cases) and CD79B (26% of cases) genes.[5] The exact mutation seen in the MYD88 (i.e. L265P)[10] and some or the mutations in CD79B[11] occur in diverse types of lymphoma. Other abnormalities seen in the small numbers of cases that have been studied so far include translocations between chromosome 14 and 18; tandem triplications of both the BCL2 gene located on the long arm of chromosome 18 at position q21 and the KMT2A gene located on the long arm of chromosome 11 between positions 22 and 25.[5] The product protein of BCL2 viz., Bcl-2, regulates cell survival and apoptosis (i.e. programmed cell death) and the product protein of KMT2a viz., MLL regulates cell maturation. Abnormalities in BCL2[12] and KMT2A[13] are associated with other types of B-cell lymphomas. It seems likely that these or other gene, chromosome, and/or gene expression abnormalities contribute to the development and/or progression of IVBCL.
The malignant B-cells in IVBCL fail to express the CD29 protein while the endothelial cells in close proximity to the intravascular accumulations of the malignant B-cells fail to express key CXC chemokine receptor proteins particularly CxcL12 but also Cxcr5, Ccr6, and/or Ccr7. The failure of the endothelial cells to express these receptor proteins may be due to the action of nearby malignant B-cells. In any event, all of the cited proteins are involved in the movement of B-cells from the intravascular space across the vascular endothelium and into tissues. The lack of these proteins may explain the accumulation of the malignant B-cells of IVLBC within blood vessels.[5]
In about 80% of cases, the malignant B-cells in IVBCL are "non-germinal center B-cells" as defined by the Hans algorithm[14] rather than the "germinal center B-cells" that are commonly found in less aggressive B-cell lymphomas. This factor may contribute to the aggressiveness of IVBCL.[5]
Presentation
Individuals presenting with the classical variant of IVLBL are typically middle-aged or elderly (39–90 years) that have one or more of the following: systemic symptoms, particularly fever (45% of cases);[5] cutaneous lesions (40%); central nervous system disorders (35%);[2] and clinical and laboratory abnormalities involving the bone marrow (~18%), lung (~6%), and, rarely, endocrine glands (e.g. pituitary, thyroid, adrenal gland[2]), liver, prostate, uterus, eye, intestine, and in individual cases almost any other organ or tissue.[15] These findings are based primarily on studies of 740 patients conducted in Europe; a study conducted in Quebec, Canada on 29 patients gave similar results.[15] Individuals may present with one, two, or more of these abnormalities. Systemic symptoms include not only the most commonly seen one viz., fever, but also malaise, weight loss, and other B symptoms; the cutaneous lesions include singular or multiple plaques, nodules, tumors, and ulcerations, some of which may be painful and most of which are located on the breast, lower abdomen, and/or extremities. Central nervous system defects include sensory and/or motor neuropathy, spinal nerve root pain, paresthesia, hypoesthesia, aphasia, dysarthria, hemiparesis, seizures, myoclonus, transient visual loss, vertigo, altered conscious states, and, particularly in relapsed disease, neurolymphomatosis (i.e. direct invasion of one or more nerves in the peripheral nervous system by the malignant B-cells).[5] Laboratory studies generally show non-specific abnormalities: elevated levels of serum lactate dehydrogenase and soluble IL2RA;[16] anemia, decreases in blood platelet levels, and decreases in white blood cell levels in 25%->50% of cases.[7] Circulating malignant B-cells are not found in 90-95% of cases[5] and laboratory evidence of organ injury is found in those cases involving these organs.[7]
Diagnosis
The diagnosis of IVBCL is heavily dependent upon obtaining biopsy specimens from involved tissues, particularly the skin but in cases without skin lesions, other apparently involved tissues. Microscopic examination of these tissues typically shows medium-sized to large-sized lymphocytes located within small- to medium-sized blood vessels of the skin, lung, and other tissues or the sinusoids of the liver, bone marrow, and spleen. On occasion, these malignant cells have the appearance of Reed-Sternberg cells. The lesions should show no or very little extension outside of blood vessels. As determined by immunohistochemistry analyses, the intravascular malignant lymphocytes express typical B-cell proteins, particularly CD20, which is found in almost all cases, CD79a and Pax5, which are found in most cases,[5] and MUM1 and Bcl-2, which are found in 95% and 91% of cases, respectively.[2] These B-cells are usually (80% of cases) non-germinal center B-cells (see Pathophysiology section) and may express one or more of the gene, chromosome, and gene expression abnormalities described in the above Pathophysiology section. Since the classical variant can present with a wide range of clinical signs, symptoms, and organ involvements, its presence may not be apparent, particularly in cases that do not exhibit clinically apparent skin lesions. Accordingly, random skin biopsies have been used to obtain evidence of IVL in cases that have signs and/or symptoms of the disease that are restricted to non-cutaneous sites,[2] even in cases that present with no other finding except unexplained fever.[17] The diagnosis of IVBCL, classical variant is solidified by finding these pathological features in more than one site.[2]
Treatment and prognosis
At diagnosis, IVBCL must be regarded as an aggressive and disseminated malignancy requiring systemic chemotherapy. In the absence of long- or short-term, controlled clinical trials on treating this lymphoma, individuals with IVBCL have been treated with the standard regimen used to treat diffuse large B-cell lymphomas viz., the CHOP chemotherapy regimen which consists of cyclophosphamide, hydroxydaunorubicin (also termed doxorubicin or adriamycin), oncovin (also termed vincristine), and a corticosteroid (i.e. either prednisone or prednisolone) plus the monoclonal antibody immunotherapy agent, Rituximab. This immunochemotherapeutic regimen has achieved an overall survival rate at 3 years of 81%; this overall survival rate using CHOP before Retuximab was added to the regimen was only 33%. However, highly toxic reactions to Rituximab such as pulmonary failure may occur and require delay or interrupting the use of this drug. High dose chemotherapy regimens followed by autologous stem-cell transplantation has offered clinical improvement similar to that found with the CHOP plus Rituximabn. However, only a small percentage of patients with IVBCL are young and healthy enough to receive this regimen.[5] Intravenous methotrexate may be a useful addition to the rituximab-CHOP regiment in individuals with central nervous system involvement.[18][19]
Presentation
The cutaneous variant, which comprises a small percentage of all IVBCL cases, occurs almost exclusively in females and younger individuals (median age 59 years) than the classical variant (median age 72 years).[5] Individuals present with lesions that are exclusively or greatly confined to the skin.[2] The clinical features of these lesions are similar to those described in the section on Presentation of the classical variant. Individuals with the cutaneous variant may have systemic symptoms but this occurs less frequently (30% of cases) than those in the classical variant (45% of cases). In general, cutaneous variant patients are in much better physical shape than those with other forms of IVBCL and have a better long-term prognosis.[5]
Diagnosis
The diagnosis of IVL, cutaneous variant depends on finding the pathological picture in the skin as described for the classical variant except that the lesions occur exclusively or predominantly in the skin. Ideally, these pathological findings should be found in more than one skin site.[5] However, cutaneous involvement is frequently detected in a single site such as the hypervascular lesions of cherry hemangiomas and angiolipomas.[20]
Treatment and prognosis
Historically, individuals with the cutaneous variant survive significantly longer than that those with the classical variant (3 year overall survival 56% versus 22%). Early intervention in the cutaneous variant would appear to be highly desirable.[5] Virtually all reports on the treatment of the cutaneous variant were made before Rituximab was used to treat IVL. Historically, patients with localized disease obtained prolonged remission with conventional CHOP therapy. However, individuals with single cutaneous lesions were long‐term survivors: when treated with just radiation therapy or surgical removal, these single-lesion patients had prolonged remissions both at initial diagnosis and after relapse. In contrast, patients with multiple lesions had a far worse outcome after treatment with CHOP: they had an objective response in 86% of cases but nonetheless the majority relapsed within a year of treatment with and only a few being successfully managed with salvage chemotherapy.[21] Rituximab may improve the latter situation.
Presentation
The hemophagocytic syndrome-associated variant of IVBCL is a very rare variant of IVBCL. Its previous name, intravascular large B-cell lymphoma, Asian variant, was recently changed to its current name by the world Health Organization, 2016. Unlike the classical and cutaneous variants, the hemophagocytic syndrome-associated variant presents with the hemophagocytic syndrome. This syndrome is characterized by bone marrow involvement, reduced numbers of circulating blood platelets[5] as well as the reduced levels of other circulating blood cells,[22] and enlarged liver and spleen. Less often, it is also associated with overt hemophagocytosis (i.e. the engulfment by non-malignant histiocytes of red blood cells, white blood cells, platelets, and their precursor cells that is most commonly found in the bone marrow and other tissues.[5] The syndrome often reflects excessive secretion of inflammatory cytokines and severe systemic inflammation similar to that seen in the cytokine storm syndrome.[2] In general, individuals present with a rapidly progressive disease (median time from onset to diagnosis~4 weeks, range 2–12 weeks).[12] Patients are often extremely ill[12] and experience multiple organ failures.[2]
Diagnosis
The diagnosis of the intravascular large B-cell lymphoma, hemophagocytic syndrome-associated variant depends on the individual presenting with clinical and laboratory findings compatible with the hemphagocytic syndrome (see previous section) and on the histology of biopsied tissues of the bone marrow, spleen, liver, brain, or other organ that clinical and/or laboratory findings suggest are involved in the disease. Its histology is described in the Diagnosis section of the classical variant but also includes the presence of hemophagocytosis, i.e. the engulfment of red blood cells and/or other mature and immature blood cells.[5] Hemophagocytosis can also be found in sites removed from the intravascular lesions such as the cerebrospinal fluid in patients with central nervous system involvement.[22]
Treatment and prognosis
Prior to the use of rituximab, individuals with this variant generally followed a rapidly (i.e. weeks to months) fatal course even when treated with the CHOP regimen. However, addition of rituximab to the CHOP regimen appears to have improved the treatment of this disease. Intravenous methotrexate may be a useful addition to the rituximab-CHOP regiment in individuals with central nervous system involvement.[18][19]
Intravascular NK/T cell lymphomas
Pathophysiology
Three studies have examine gene mutations and gene expression abnormalities in IVNK/TL. A retrospective study of 25 patients identified numerous gene abnormalities including tumor-specific splicing alterations in oncogenes and tumor suppressor genes such as HRAS, MDM2, and VEGFA as well as premature termination mutations or copy number losses in a total of 15 splicing-regulator genes such as SF3B5 and TNPO3.[23] A study of two patients with IVNKL identified mutations in genes that produce histone proteins (HIST1H2BE, HIST1H2BN and H3F3A), the histone deacetylase gene, HDAC5, two genes that produce helicase proteins (WRN and DDX3X), two genes that make DNA methylation-related enzymes (TET2 and DNMT1) and a gene in the SWI/SNF family of chromatin remodeling genes, ARID1A.[24] In a third study of a single patient, copy number analysis identified driver gene alterations in ARID1B, HACE1, and SMAD4 genes and gain of the SOX2 gene.[8] While further studies are needed before conclusions can be made, one or more of these gene abnormalities may contribute to the development and/or progression of IVNK/TL.
The malignant NK and T cells that accumulate within the vascular of individuals with IVNK/TL are usually infected with the Epstein–Barr virus (EBV). This suggests that most IVNK/TL cases are examples of the Epstein-Barr virus-associated lymphoproliferative diseases and, like these diseases, are EBV-driven.[8] About 95% of the world's population is infected with EBV. During the initial infection, the virus may cause infectious mononucleosis, only minor non-specific symptoms, or no symptoms. Regardless of this, the virus enters a latency phase and the infected individual becomes a lifetime asymptomatic carrier of EBV. Weeks, months, years, or decades thereafter, a small percentage of these carriers develop an EBV-associated lymphoproliferative disease,[25][26] including in extremely rare cases IVNK/TL.[8] EBV is well known to infect NK- and T-cells, to express some of its genes that promote the survival and proliferation of the cells it infects, and thereby to cause various and far more common NK- and T-cell lymphomas.[27] It seems likely that the virus acts similarly to cause IVNK/TL.[8] IVNK/TL may differ from the other types of NK- and T-cell lymphomas which EBV produces because its NK- and T-cells and nearby endothelial cells have defects in the expression of proteins required for the NK/T-cells to pass through the endothelium and into the surrounding tissues (see above section on the Pathopysiology IVBCL).[28]
Presentation
Individuals (age range 23–81 years[8]) with IVNK/TL typically have a rapidly progressive disease. They commonly present with skin lesions, less commonly symptoms due to central nervous system involvement, and in a minority of cases symptoms due to the involvement of the bone marrow, liver, kidneys, ovaries, and/or cervix.[1] They often show signs of an disseminated disease such as fever, weight loss, night sweats, arthralgias, jaundice, decreased numbers of circulating red blood cells, white blood cells, and/or platelets, bone marrow involvement as determined by biopsy, and signs/symptoms of multiple organ involvement.[8]
Diagnosis
The diagnosis of IVNK/TL depends upon obtaining histology findings in the skin and/or other involved tissue that resembles that seen in IVBCL except that the malignant lymphocytes are not B-cells but rather: 1) NK-cells as evidenced by their expression of NK-cell selective marker proteins (e.g. CD3e, CD2, CD30, CD43, CD56, and/or CD79), expression of granule-bound enzymes (e.g. granzyme B),[8] and expression of EBV proteins (e.g. Epstein–Barr virus latent membrane protein 1[8] and EBV-produced small RNAs[1]); but not the expression of B-cell (e.g. CD20, CD79a, and Pax5) or cytotoxic T cell marker proteins;[8] and 2) cytotoxic T-cell lymphoma as evidenced by the neoplastic cells' expression of T-cell co-receptor proteins (e.g. CD3, CD4, and/or CD8) as well as EBV marker proteins and/or small RNAs but usually not B-cell or NK-cell marker proteins.[9]
Treatment and Prognosis
Patients with IVNK/TL have been treated with various chemotherapy regimens, particularly CHOP or, less commonly, hyperCVAD. Rare patients have been treated with chemotherapy followed by hematopoietic stem cell transplantation or chemotherapy plus a proteasome inhibitor, bortezomib. In general, patients have responded poorly to treatment, have short (i.e. up to 12 months) survival times regardless of the chemotherapy regimen used.[8][9][29][30] Rituximab does not target NK- or T-cells and therefore is not used to treat IVNK/TL.
References
- Bi Y, Huo Z, Liang Z, Meng Y, Jia C, Shi X, Song L, Luo Y, Ling Q, Liu T (July 2015). "Intravascular NK-cell lymphoma: a case report and review of the literature". Diagnostic Pathology. 10: 84. doi:10.1186/s13000-015-0336-7. PMC 4488042. PMID 26126576.
- Korkolopoulou P, Vassilakopoulos T, Milionis V, Ioannou M (July 2016). "Recent Advances in Aggressive Large B-cell Lymphomas: A Comprehensive Review". Advances in Anatomic Pathology. 23 (4): 202–43. doi:10.1097/PAP.0000000000000117. PMID 27271843. S2CID 205915174.
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES (May 2016). "The 2016 revision of the World Health Organization classification of lymphoid neoplasms". Blood. 127 (20): 2375–90. doi:10.1182/blood-2016-01-643569. PMC 4874220. PMID 26980727.
- Yan J, Zhang F, Luo D, Yao S, Chen Y, Xu F, Luo X, He J, Liu Y (2017). "Intravascular NK/T-cell lymphoma: a series of four cases". International Journal of Clinical and Experimental Pathology. 10 (9): 9541–9550. PMC 6965900. PMID 31966830.
- Ponzoni M, Campo E, Nakamura S (October 2018). "Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks". Blood. 132 (15): 1561–1567. doi:10.1182/blood-2017-04-737445. PMID 30111607.
- PFLEGER L, TAPPEINER J (August 1959). "[On the recognition of systematized endotheliomatosis of the cutaneous blood vessels (reticuloendotheliosis?]". Der Hautarzt; Zeitschrift für Dermatologie, Venerologie, und Verwandte Gebiete (in German). 10: 359–63. PMID 14432547.
- Shimada K, Kinoshita T, Naoe T, Nakamura S (September 2009). "Presentation and management of intravascular large B-cell lymphoma". The Lancet. Oncology. 10 (9): 895–902. doi:10.1016/S1470-2045(09)70140-8. PMID 19717091.
- Zanelli M, Mengoli MC, Del Sordo R, Cagini A, De Marco L, Simonetti E, Martino G, Zizzo M, Ascani S (November 2018). "Intravascular NK/T-cell lymphoma, Epstein-Barr virus positive with multiorgan involvement: a clinical dilemma". BMC Cancer. 18 (1): 1115. doi:10.1186/s12885-018-5001-6. PMC 6238309. PMID 30442097.
- Gleason BC, Brinster NK, Granter SR, Pinkus GS, Lindeman NI, Miller DM (February 2008). "Intravascular cytotoxic T-cell lymphoma: A case report and review of the literature". Journal of the American Academy of Dermatology. 58 (2): 290–4. doi:10.1016/j.jaad.2006.12.022. PMID 18222325.
- Weber AN, Cardona Gloria Y, Çınar Ö, Reinhardt HC, Pezzutto A, Wolz OO (November 2018). "Oncogenic MYD88 mutations in lymphoma: novel insights and therapeutic possibilities". Cancer Immunology, Immunotherapy. 67 (11): 1797–1807. doi:10.1007/s00262-018-2242-9. PMID 30203262. S2CID 52186614.
- Cetin GO, Baris IC, Caner V, Sarikepe B, Sen Turk N, Tepeli E, Hacioglu S, Sari I, Bagci G, Keskin A (March 2016). "Mutational status of EZH2 and CD79B hot spots in mature B-cell non-Hodgkin's lymphomas: novel CD79B variations have been revealed". European Review for Medical and Pharmacological Sciences. 20 (5): 830–6. PMID 27010137.
- Li S, Lin P, Medeiros LJ (August 2018). "Advances in pathological understanding of high-grade B cell lymphomas". Expert Review of Hematology. 11 (8): 637–648. doi:10.1080/17474086.2018.1494567. PMID 29989509. S2CID 51606586.
- Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA (June 2005). "Global and Hox-specific roles for the MLL1 methyltransferase". Proceedings of the National Academy of Sciences of the United States of America. 102 (24): 8603–8. Bibcode:2005PNAS..102.8603G. doi:10.1073/pnas.0503072102. PMC 1150839. PMID 15941828.
- Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Müller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, Pan Z, Farinha P, Smith LM, Falini B, Banham AH, Rosenwald A, Staudt LM, Connors JM, Armitage JO, Chan WC (January 2004). "Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray". Blood. 103 (1): 275–82. doi:10.1182/blood-2003-05-1545. PMID 14504078.
- Fonkem E, Lok E, Robison D, Gautam S, Wong ET (August 2014). "The natural history of intravascular lymphomatosis". Cancer Medicine. 3 (4): 1010–24. doi:10.1002/cam4.269. PMC 4303169. PMID 24931821.
- Sukswai N, Lyapichev K, Khoury JD, Medeiros LJ (January 2020). "Diffuse large B-cell lymphoma variants: an update". Pathology. 52 (1): 53–67. doi:10.1016/j.pathol.2019.08.013. PMID 31735345.
- di Fonzo H, Contardo D, Carrozza D, Finocchietto P, Rojano Crisson A, Cabral C, de Los Angeles Juarez M (May 2017). "Intravascular Large B Cell Lymphoma Presenting as Fever of Unknown Origin and Diagnosed by Random Skin Biopsies: A Case Report and Literature Review". The American Journal of Case Reports. 18: 482–486. doi:10.12659/ajcr.903816. PMC 5421743. PMID 28461685.
- Komeno Y, Akiyama M, Okochi Y, Tokuda H, Abe K, Iihara K, Ryu T (2019). "Hemophagocytic Syndrome-Associated Variant of Methotrexate-Associated Intravascular Large B-Cell Lymphoma in a Rheumatoid Arthritis Patient". Case Reports in Hematology. 2019: 8947616. doi:10.1155/2019/8947616. PMC 6755279. PMID 31612088.
- Yang JJ, Chen XC, Tang Y, Shen K, Xie LP, Liu T (January 2018). "[Intravascular Large B-cell Lymphoma: a Clinical Analysis of 17 Cases]". Sichuan da Xue Xue Bao. Yi Xue Ban = Journal of Sichuan University. Medical Science Edition (in Chinese). 49 (1): 145–147. PMID 29737107.
- Saito T, Matsuya T, Takahashi C, Kaneta K, Ohishi Y, Uehara J, Hashimoto M, Honma M, Ishida-Yamamoto A (March 2017). "Cutaneous variant of intravascular large B-cell lymphoma in a Japanese patient: An occult lesion localized in a solitary angiolipoma". The Journal of Dermatology. 44 (3): e28–e29. doi:10.1111/1346-8138.13504. PMID 27422850.
- Ferreri AJ, Campo E, Seymour JF, Willemze R, Ilariucci F, Ambrosetti A, Zucca E, Rossi G, López-Guillermo A, Pavlovsky MA, Geerts ML, Candoni A, Lestani M, Asioli S, Milani M, Piris MA, Pileri S, Facchetti F, Cavalli F, Ponzoni M (October 2004). "Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the 'cutaneous variant'". British Journal of Haematology. 127 (2): 173–83. doi:10.1111/j.1365-2141.2004.05177.x. PMID 15461623. S2CID 19736970.
- Verma A, Sharma A, Robetorye R, Porter A, Hilal T (2020). "Intravascular Large B-cell Lymphoma Associated with Systemic and Central Nervous System Hemophagocytic Lymphohistiocytosis: A Case Report". The Permanente Journal. 24. doi:10.7812/TPP/19.105. PMC 6907904. PMID 31852057.
- Fujikura K, Yoshida M, Uesaka K (March 2020). "Transcriptome complexity in intravascular NK/T-cell lymphoma". Journal of Clinical Pathology. 73 (10): 671–675. doi:10.1136/jclinpath-2020-206461. PMID 32188628. S2CID 213186240.
- Fujikura K, Yamashita D, Sakamoto R, Ishikawa T, Chuang SS, Itoh T, Imai Y (September 2019). "Intravascular NK/T-cell lymphoma: clinicopathological and integrated molecular analysis of two cases provides a clue to disease pathogenesis". Journal of Clinical Pathology. 72 (9): 642–646. doi:10.1136/jclinpath-2019-205727. PMID 31123138. S2CID 163167895.
- Houldcroft CJ, Kellam P (March 2015). "Host genetics of Epstein–Barr virus infection, latency and disease". Reviews in Medical Virology. 25 (2): 71–84. doi:10.1002/rmv.1816. PMC 4407908. PMID 25430668.
- Farrell PJ (August 2018). "Epstein–Barr Virus and Cancer". Annual Review of Pathology. 14: 29–53. doi:10.1146/annurev-pathmechdis-012418-013023. PMID 30125149. S2CID 52051261.
- de Mel S, Soon GS, Mok Y, Chung TH, Jeyasekharan AD, Chng WJ, Ng SB (June 2018). "The Genomics and Molecular Biology of Natural Killer/T Cell Lymphoma: Opportunities for Translation". International Journal of Molecular Sciences. 19 (7): 1931. doi:10.3390/ijms19071931. PMC 6073933. PMID 29966370.
- Alhumidi A (July 2015). "Cutaneous Intravascular NK/T-cell lymphoma mimic panniculitis clinically, case report and literature brief review". Diagnostic Pathology. 10: 107. doi:10.1186/s13000-015-0330-0. PMC 4504160. PMID 26178620.
- Wang L, Chen S, Ma H, Shi D, Huang C, Lu C, Gao T, Wang G (September 2015). "Intravascular NK/T-cell lymphoma: a report of five cases with cutaneous manifestation from China". Journal of Cutaneous Pathology. 42 (9): 610–7. doi:10.1111/cup.12515. PMID 25931234. S2CID 23046075.
- Melchers RC, Willemze R, Jansen PM, Daniëls LA, Vermeer MH, Quint KD (June 2019). "A rare case of cutaneous Epstein-Barr virus-negative intravascular cytotoxic T-cell lymphoma". JAAD Case Reports. 5 (6): 548–551. doi:10.1016/j.jdcr.2019.04.013. PMC 6581970. PMID 31245517.