Proteases in angiogenesis
Angiogenesis is the process of forming new blood vessels from existing blood vessels, formed in vasculogenesis. It is a highly complex process involving extensive interplay between cells, soluble factors, and the extracellular matrix (ECM). Angiogenesis is critical during normal physiological development, but it also occurs in adults during inflammation, wound healing, ischemia, and in pathological conditions such as rheumatoid arthritis, hemangioma, and tumor growth.[1][2] Proteolysis has been indicated as one of the first and most sustained activities involved in the formation of new blood vessels. Numerous proteases including matrix metalloproteinases (MMPs), a disintegrin and metalloproteinase domain (ADAM), a disintegrin and metalloproteinase domain with throbospondin motifs (ADAMTS), and cysteine and serine proteases are involved in angiogenesis. This article focuses on the important and diverse roles that these proteases play in the regulation of angiogenesis.
MMPs
Matrix metalloproteinases (MMPs) are a large multigene family of zinc-dependent endopeptidases. The collective MMP family is capable of degrading all known ECM macromolecules. MMP activity is regulated at the level of transcription, post-translationally by proteolytic cleavage, and by endogenous inhibitors known as tissue inhibitors of metalloproteinases (TIMPs). The role of matrix metalloproteinases and TIMPs in several pathological conditions including angiogenesis, tumor growth, and metastasis has been investigated and very well described.
Matrix metalloproteinases contain five conserved domains/sequence motifs:
- Signal peptide sequence, which directs the enzyme into the rough endoplasmic reticulum during synthesis
- Propeptide domain, which is cleaved to activate the enzyme
- Catalytic domain, which contains the conserved Zn2+ binding region and mediates enzyme activity
- Hemopexin domain, which provides the substrate specificity
- Small hinge region, which allows the hemopexin domain to bring the substrate to the active core of the catalytic domain
There is also a subfamily of the matrix metalloproteinases, the membrane-type MMPs (MT-MMPs) which contain an additional transmembrane domain and a short cytoplasmic domain. After activation of MMPs by removal of the propeptide domain, their activity is regulated by TIMPs. TIMPs specifically and reversibly inhibit the activity of MMPs. So far there have been identified four members of the family, TIMP1–4. All TIMPs contain twelve conserved cystein residues, which form six disulfide bonds. The C-terminal domains of TIMPs are highly variable and confer their specificity towards preferred MMP targets.[3][4]
ADAM/ADAMTS
ADAMs comprise a family of integral membrane as well as secreted glycoproteins which are related to snake venom metalloproteinases and MMPs. Like MMPs, ADAMs are composed of multiple conserved domains. They contain propeptide, metalloproteinase, disintegrin-like, cystein-rich, and epidermal growth factor like domains, although variations in domain composition have been observed in non-animal organisms.[5] Membrane anchored ADAMs contain a transmembrane and cytoplasmic domain. The domains contained within the ADAMs family have been characterized, uncovering their functional and structural roles.[6] ADAMs contain a consensus sequence which has three histidine residues that bind to the catalytically essential zinc ion. The propeptide is removed through cleavage by a furin type protease yielding the active enzyme. The propeptide of most MMPs is cleavable by proteases such as trypsin, plasmin, chymotrypsin and other MMPs.[7] ADAMs participate in a wide variety of cell surface remodeling processes, including ectodomain shedding, regulation of growth factor availability and mediating cell-matrix interactions. ADAM17 and ADAM15 have recently been identified in endothelial cells (EC).[8]
ADAMTS are a subfamily of ADAM related metalloproteinases that contain at least one thrombospondin type I sequence repeat motif (TSR). They are secreted proteins; and the TSR facilitates their localization to the ECM placing it in close proximity to their substrates. Functionally, ADAMTS can be divided into three groups: procollagen aminopeptidase, aggrecanase, and ADAMTS13 which cleaves von Willebrand factor. Unlike with MMPs, TIMPs are more selective in their ability to inhibit ADAMs and ADAMTSs. TIMP3 is able to inhibit ADAM17 and 12 as well as ADAMTS4 and 5. ADAM8 and ADAM9 are not susceptible to inhibition by TIMPs.
Other proteolytic enzymes
Many additional classes of enzymes have been identified that facilitate angiogenesis. They include serine, aspartic, and cysteine-type proteases. A highly characterized example of the serine protease family is the plasminogen activator-plasmin system, which has been shown to be involved in vascular remodelling . Tissue plasminogen activator (tPA), and urokinase plasminogen activator (urokinase, uPA) are serine proteases which cleave and activate plasminogen. The activated form of plasminogen, plasmin, is a wide-ranging protease capable of acting on various ECM components including fibrin, collagens, laminin, fibronectin, and proteoglycans.[9] Additionally, plasmin also is able to activate various other MMPs.
In humans, the group of cathepsin cysteine proteases or cysteine cathepsins comprises 11 family members, cathepsins B, C, F, H, L1, L2, K, O, S, W, and X/Z.[10] Cysteine cathepsins are synthesized as inactive zymogens and activated by proteolytic removal of their propeptide. These enzymes are primarily localized in lysosomes and function in terminal protein degradation and processing. Cathepsins also can be secreted by cells, associate with the cell surface, and degrade the ECM. A study of all 11 members of the cathepsin family highlights their importance in tumorigenesis and tumor associated angiogenesis.[11] Examination of cathepsin activity by using chemical probes and in vivo imaging techniques demonstrated an increase in cathepsin activity in the angiogenic blood vessels and invasive fronts of carcinoma in the RIP-Tag2 transgenic mouse model of pancreatic islet tumor genesis.
Aminopeptidases function as exopeptidases which remove amino acids from the amino-terminus of proteins. Aminopeptidase N (CD13/APN) is highly expressed on the endothelium of growing vessels.[12] Inhibitors of CD13/APN dramatically impair tumor growth.
Ectodomain shedding
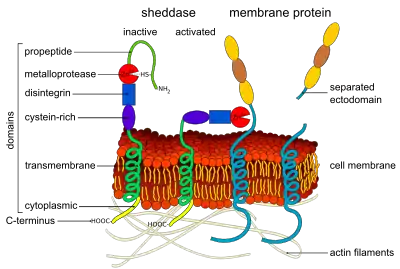
It has become clear in the past years that ectodomain shedding is an initial step for the activation of specific receptors such as Notch, ErbB-4 and the angiopoietin receptor Tie-1. Notch-1 signaling is essential for endothelial differentiation, and tumor angiogenesis, while the angiopoietin receptor Tie-1 facilitates embryonic blood vessel formation.[13][14] Upon binding of their ligands, Notch-1 and Tie-1 undergo proteolytic cleavage of the ectodomains by ADAM17 and ADAM10. This cleavage frees the cytoplasmic fragment for cellular signaling. In the case of Notch-1, it transfers to the nucleus.
Many cytokines and growth factors are synthesized as membrane-bound proforms which undergo proteolytic shedding for activation. The ephrins EPH receptor A2 and A3 are shed by ADAM10, creating cleaved soluble Eph receptors, which inhibit tumor angiogenesis in mice.[15] Additional examples are the proteolytic shedding of soluble E-selectin,[16] shedding of urokinase receptor (uPAR) by MMP-12 creating soluble uPAR which has chemotactic properties for leukocytes and progenitor cells, and the shedding of interleukin-6 receptors by ADAM10 and ADAM17 which facilitates interleukin-6 signaling in endothelial cells.[17] Semaphorin 4D is cleaved from its membrane-bound form by MT1-MMP (MMP-14) in tumor cells; it then interacts with plexin B1 on endothelial cells, promoting pro-angiogenic chemotaxis.[18] Shedding of a membrane-anchored cytokine or growth factor by ADAM proteinases may be relevant for various signal transduction events. Alternatively, shedding may be required for the ligand to diffuse to distant receptors. Shedding may be required for the down regulation of signals by removing signaling ligands, or cleavage and release of receptors. Release of the receptor may also generate soluble receptors which act as decoys by sequestering ligands. These findings indicate that ectodomain shedding is a ubiquitous process facilitating a wide variety of cellular events involved in angiogenesis. Because potent biological modifiers are generated, it is likely controlled by highly regulated mechanism. Along with ADAMs and MT-MMPs, membrane-bound serine proteases also may play a role in ectodomain shedding.
Proteolytic degradation of the extracellular matrix (ECM)
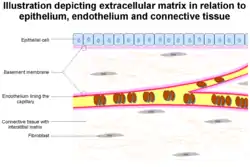
The formation of capillaries from pre-existing blood vessels requires the remodeling of both the peicapillary membrane of the parent venule, as well as the local and distal ECM. At the onset of angiogenesis endothelial cells (EC) must remodel three different barriers in order to migrate and invade the target tissue. First is the basement membrane between the endothelium and vascular smooth muscle cells or pericytes, followed by the fibrin gel formed from fibrinogen that is leaked from the vasculature, and finally the extracellular matrix in the target tissue. The vascular basement membrane is composed of type IV collagen, type XV collagen, type XVIII collagen, laminins, entactin, heparan sulfate proteoglycans, perlecan, and osteonectin. All of these components of the basement membrane are substrates for MMP-2, 3, 7, and 9, among others. Inhibitors of MMP activity have spotlighted the importance of these proteins in controlling angiogenesis. Recently, it has been discovered that small interfering RNA (siRNA) mediated target RNA degradation of urokinase receptor and MMP-9 inhibits the formation of capillary like structures in both in vitro and in vivo models of angiogenesis.[19] After working their way through the basement membrane, EC must invade through a dense fibrin gel which is polymerized from fibrinogen derived from the vascular bed.[20] Plasmin, an effective fibrinolysin produced by tPA or uPA, was thought to be essential in this process, but plasminogen deficient mice do not display major defects of neovascularization in fibrin rich tissues.[21] These findings highlight the diverse amount of proteolytic enzymes ECs use to remodel the ECM. For example, MMP-3, 7, 8, 12 and 13 can cleave fibrinogen.[22]
MMP activity is one of the earliest and most sustained processes that take place during angiogenesis. By studying the transition from an avascular to a vascular tumor Fang et al. were able to identify the key role of MMP-2 in angiogenesis. MMP-2 expression and activity was increased in angiogenic tumors as compared with avascular tumors, and the addition of antisense oligonucleotides targeting MMP-2 inhibits the initiation of angiogenesis maintaining the avascular phenotype. This data along with other reports suggest that MMP activity is necessary to initiate the earliest stages of angiogenesis and tumor development. The creation of MMP deficient mice has provided important insight into the role of MMPs in the regulation of angiogenesis. For example, MMP-2 knockout mice develop normally but display significant inhibition of corneal angiogenesis.[23]
Proteolytic fragments as regulators of angiogenesis
Numerous proteolytic fragments or domains of ECM proteins have been reported to exert positive or negative activity on angiogenesis. Native proteins which contain such domains with regulatory activity are normally inactive, most likely because they are cryptic segments hidden in the native protein structure. Angiostatin is a 38 kDa plasminogen fragment with angiogenesis inhibitor activity. Angiostatin fragments contain kringle domains which exert their inhibitory activity at several different levels; they inhibit endothelial cell migration and proliferation, increase apoptosis, and modulate the activity of focal adhesion kinase (FAK). Endostatin is a 20 kDa fragment of collagen XVIII. The major role of endostatin is in its ability to potently inhibit endothelial cell migration and induce apoptosis.[24] These effects are mediated by interacting and interfering with various angiogenic related proteins such as integrins and serine/threonine-specific protein kinases. Numerous studies have demonstrated that tropoelastin, the soluble precursor of elastin, or proteolytic elastin fragments have diverse biological properties. Nackman et al. demonstrated that elastase generated elastin fragments mediate several characteristic features of aneurysmal disease which correlated to angiogenesis. Osteonectin is a metal binding glycoprotein produced by many cell types including ECs. Lastly, endorepellin is a recently described inhibitor of angiogenesis derived from the carboxy terminus of perlecan.[25] Nanomolar concentrations of endorepellin inhibits EC migration and angiogenesis in different in vitro and in vivo models by blocking EC adhesion to various substrate such as fibronectin and type I collagen.
Endogenous inhibitors or activators generated by proteolytic degradation of larger proteins mostly from the ECM have proven to contribute to the regulation of tumor growth and angiogenesis. This article mentions only a small fraction of the known proteolytic fragments which alter EC behavior and function during angiogenesis. This abundance has garnered increased attention because of their potential for anti-angiogenic and anti-cancer therapies.
Proteolytic activation of growth factors
Proteases not only modulate cell-matrix interactions but also can control the onset and progression of angiogenesis by activating angiogenic growth factors and cytokines. Hepatocyte growth factor (HGF), an angiogenesis promoting growth factor, is activated by HGF activation factor, a serine protease related to plasminogen.[26] Several growth factors such as basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) are trapped in the ECM by various proteoglycans. The proteolytic degradation of these proteoglycans liberates the growth factors allowing them to reach their receptors and influence cellular behavior. Growth factors that indirectly affect angiogenesis are also targets of proteolytic activation. For example, plasminogen activators drive the activation of latent transforming growth factor beta (TGF-β) from bone ECM and thus modulate angiogenesis in bone.[27]
Proteases not only have the ability change the availability of growth factors, but can also modify their properties. This ability was shown for VEGF165 that is cleaved by MMP-3 or MMP-9 to a smaller molecule with properties similar to VEGF121.[28] These two isoforms of VEGF have very different properties. VEGF165 induces a regular vessel pattern during tumor neovascularization. VEGF121 and the truncated VEGF165, in contrast, cause irregular patterns of neovascularization, most likely due to their inability to bind heparan sulfates, wherefore they do not provide any spatial information that is buried in the ECM. Another important factor in angiogenesis, stromal cell-derived factor-1 (SDF-1), is also modified by the aminodipeptidase dipeptidyl peptidase-4 (DPP4). Cleavage of SDF-1 reduces it heparan sulfate affinity and interactions with its receptor CXCR4 are reduced.[29] The ADAM family of proteases is receiving increased attention for their ability to alter the balance between pro-and anti-angiogenic factors. ADAM17 is able to release active tumor necrosis factor-alpha (TNFα) and heparin-binding EGF-like growth factor (HB-EGF) from their membrane bound precursors which can indirectly affect angiogenesis.[30]
Proteases as inhibitors of angiogenesis
Proteases not only facilitate angiogenesis, but they also have the ability to put the brakes on the process. One example of this is the processing of angiogenesis inhibitors by MMPs. As previously described, MMPs have been shown to cleave plasminogen and collagen XVIII into the endogenous angiogenesis inhibitors angiostatin and endostatin. MMP-2 itself possesses anti-angiogenic properties that are independent of its catalytic domain. Interactions between integrin αvβ3 and MMP-2 on the EC cell surface may be necessary for MMP-2 activity during angiogenesis. The hemopexin like domain in the carboxy terminus of MMP-2 is able to block this interaction of active MMP-2 and integrin αvβ3 on the EC surface which lead to inhibition of MMP-2 activity.[31]
During angiogenesis ADAM15 is preferentially expressed on EC. ADAM15 is able to interact with integrins αvβ3 and α5β1 through its disintegrin domain via an RGD (arginine-glycine-aspartic acid) motif. Most disintegrins contain this conserved RGD motif, but ADAM15 is the only member of the ADAM family to contain this motif. A recombinant disintegrin domain of human ADAM15 inhibits a variety of EC functions in vitro including proliferation, adhesion, migration, and capillary formation.[32] Overexpression of ADAM15 disintegrin domain resulted in inhibition of angiogenesis, tumor growth, and metastasis. On the other hand, it has not been shown whether full length ADAM15 plays an inhibitory role in vivo. ADAMTS1 and ADAMTS8 inhibit angiogenesis in vitro in two functional angiogenesis assays. Both enzymes inhibit bFGF induced vascularization in the corneal pocket assay and inhibit VEGF induced angiogenesis in the chorioallantoic membrane assay.[33] All together, these data indicate that proteases can function as both positive and negative regulators of angiogenesis.
Proteolysis and cell migration
Angiogenesis requires the migration and invasive growth of cells. This is facilitated by a balanced interplay between detachment and formation of cell adhesions which enable the cell to crawl forward through the ECM.[34] The cell uses limited proteolytic activity at sites of individual focal adhesions via the formation of multiprotein complexes. Multiprotein complexes are localized in lipid rafts on the cell surface, where membrane bound proteases are often incorporated. For example, leukocytes complex urokinase (uPA), urokinase receptor (uPAR), and integrins which participate in cell adhesion and invasion.[35] In these complexes, uPAR acts as an organizing center forming noncovalent complexes with integrins, LRP-like proteins, and urokinase. Similar complexes also are found on ECs.
Uncontrolled proteolysis of the ECM
The proteolytic activities that take place during angiogenesis require precise spatial and temporal regulation. If not for this control excessive proteolysis could lead to damage of the tissue and the loss of anchorage points for migrating cells. This is illustrated by mice which are deficient for plasminogen activator inhibitor-1 (PAI-1).[36][37] PAI-1 inhibits plasminogen activators and thus plasmin activation; therefore it could be assumed that PAI-1 deficiency would increase angiogenesis and tumor growth. Unexpectedly, when PAI-1 deficient mice were challenged with cancer cells on a collagenous matrix, angiogenesis and vascular stabilization was inhibited, hampering tumor growth. This finding was credited to the protective properties PAI-1 imparts against excessive degradation of the surrounding ECM by plasmin. Without this protection the footholds used by endothelial cells to migrate and form capillary structures are destroyed. Uncontrolled proteolysis also is attributed to the disruption of vascular development and premature deaths in murine embryos deficient of the inhibitor reversion-inducing-cysteine-rich protein with kazal motifs (RECK). This is most likely due to uncontrolled MMP activity, because a partial rescue was obtained by simultaneously knocking out RECK and MMP-2.[38]
Proteases involved in the recruitment of bone marrow derived cells during angiogenesis
Leukocytes and endothelial progenitor cells (EPCs) contribute to the initiation and guidance of new blood vessels.[39] Monocytes produce a variety of pro-angiogenic factors. There is also a population of CD34 positive cells that can express endothelial associated proteins, such as VE-cadherin and kinase insert domain receptor (KDR, VEGF receptor 2) which aid in influencing the progression of angiogenesis.[40] The absence or dysfunction of these cells is implicated in impaired vascularization in cardiac and diabetes patients.[41] MMP-9 plays a key role in mobilizing EPCs from the bone marrow. Heissig et al. have proposed a mechanism for how MMP-9 facilitates the availability of EPCs for angiogenesis. First, circulating VEGF induces MMP-9 expression in the bone marrow, MMP-9 then is able to cleave and release c-kit ligand. Activated c-kit is then able to recruit hematopoietic, endothelial and mast cell progenitor cells, these cells are then accumulated in the angiogenic area and produce large amounts of VEGF tipping the scales in favor of angiogenesis.[42]
MMP-9 is not the only protease shown to be involved in EPC enhanced angiogenesis. Cathepsin L1 is active at neutral pH by associating with a p41 splice variant of the MHC class II-associated invariant chain which is strongly expressed in EPCs.[43] This ability to stay active at neutral pH may facilitate EPC invasion, remodeling of matrix collagens and gelatin, and neovascularization. Knock out of cathepsin L1 in mice exhibited impaired blood flow restoration in ischemic limbs, indicating impaired neovascularization. Neovascularization also is impaired in mice treated with bone marrow derived cells deficient of cathepsin L1 as compared with wild type cells. The target by which cathepsin L1 stimulates angiogenesis is not yet identified.
Maturation of newly formed blood vessels via proteases
It has been well established that smooth muscle-like pericytes play an important role in stabilizing newly formed blood vessels. Pericytes present in the stroma of tumors of breast cancer patients express MMP-9.[44] Animal models deficient of MMP-9 display disturbed recruitment of pericytes.[45] The inability to recruit pericytes severely affects the stability of vessels and the degree of vascularization of neuroblastomas. Aminopeptidase A also may be involved in pericyte recruitment due to its increased expression by activated pericytes in various pathological conditions associated with angiogenesis.[46] The mechanism by which this protease facilitates vessel maturation has not yet been determined. Angiogenesis requires a fine balance between proteolytic activity and proteinase inhibition. Pericytes secrete TIMP-3 which inhibits MT1-MMP dependent MMP-2 activation on endothelial cell, thus facilitating stabilization of newly formed microvessels. Co-cultures consisting of pericytes and endothelial cells induce the expression of TIMP-3 by pericytes, while endothelial cells produce TIMP-2.[47] Together, these inhibitors stabilize the vasculature by inhibiting a variety of MMPs, ADAMs, and VEGF receptor 2.
Immature vessels remain dependent on continuous exposure the angiogenic growth factors without pericyte coverage.[48] As the reservoir of growth factors is removed the endothelial cells do not survive, and undergo caspases induced apoptosis, while other proteases participate in the degradation and removal of the remaining cell debris.
Perspective
Proteases play numerous roles in angiogenesis, both in development and especially in pathological conditions. Because they are important regulators of tissue degradation and cell migration, it is expected that their inhibition would be beneficial for inhibiting tumor growth and vascularization. Promising results have been observed in animal studies, but clinical trials have failed to demonstrate similar results and are often accompanied by unacceptable side effects.[49] This has influenced continued research which has identified new families of proteases, such as ADAM, ADAMTS, and MT-MMPs. Perhaps more significantly, a new paradigm has emerged for proteases being essential for modulating growth factors and cytokines, generating biologically active fragments from the matrix, facilitating recruitment of bone marrow derived cells, and stabilization of mature blood vessels. Better understanding of the various activities of proteases and their inhibitors will aid in more tailor made treatments for numerous disorders.
References
- Hublica, O; Brown, M; Egginton, S (1992). "Angiogenesis in skeletal and cardiac muscle". Physiol. Rev. 72 (2): 369–417. doi:10.1152/physrev.1992.72.2.369. PMID 1372998.
- Hanahan, D; Folkman, J (1996). "Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis". Cell. 86 (3): 353–364. doi:10.1016/S0092-8674(00)80108-7. PMID 8756718.
- Hessing, B; Hattori, K; Friedrich, M; Rafii, S; Werb, Z (2003). "Angiogenesis: vascular remodeling of the extracellular matrix involves metalloproteinases". Curr. Opin. Hematol. 10 (2): 136–141. doi:10.1097/00062752-200303000-00007. PMID 12579040. S2CID 25978201.
- Murphy, G; Willenbrock, F (1995). "Tissue inhibitors of matrix metalloendopeptidases". Proteolytic Enzymes: Aspartic and Metallo Peptidases. Methods in Enzymology. Vol. 248. pp. 496–510. doi:10.1016/0076-6879(95)48032-3. ISBN 9780121821494. PMID 7674941.
- Souza J, Lisboa A, Santos T, Andrade M, Neves V, Teles-Souza J, Jesus H, Bezerra T, Falcão V, Oliveira R, Del-Bem L (2020). "The evolution of ADAM gene family in eukaryotes". Genomics. 112 (5): 3108–3116. doi:10.1016/j.ygeno.2020.05.010. PMID 32437852. S2CID 218832838.
- Stone, A; Kroeger, M; Sang, QX (1999). "Structure-function analysis of the ADAM family of diintegrin-like and metalloproteinase-containing proteins". J. Protein Chem. 18 (4): 447–465. doi:10.1023/A:1020692710029. PMID 10449042. S2CID 45601048.
- Nagase, H; Woessner Jr, JF (1999). "Matrix metalloproteinases". J. Biol. Chem. 274 (31): 21491–21494. doi:10.1074/jbc.274.31.21491. PMID 10419448.
- Herren, B; Raines, EW; Ross, R (1997). "Expression of a disintegrin-like protein in cultured human vascular cells and in vivo". FASEB J. 11 (2): 173–180. doi:10.1096/fasebj.11.2.9039960. PMID 9039960. S2CID 9206855.
- Saksela, O (1985). "Plasminogen activation and regulation of pericellular proteolysis". Biochim. Biophys. Acta. 823 (1): 35–65. doi:10.1016/0304-419x(85)90014-9. PMID 2413894.
- Turk, V; Turk, B; Turk, D (2003). "Lysosomal cysteine proteases: facts and opportunities". EMBO J. 20 (17): 4629–4633. doi:10.1093/emboj/20.17.4629. PMC 125585. PMID 11532926.
- Joyce, J; Baruch, A; Chehade, K; Meyer-Morse, N; Giraudo, E; Tsai, FY; Greenbaum, DC; Hager, JH; et al. (2004). "Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis". Cancer Cell. 5 (5): 443–453. doi:10.1016/S1535-6108(04)00111-4. PMID 15144952.
- Pasqualini, R; Koivunen, E; Kain, R; Lahdenranta, J; Sakamoto, M; Stryhn, A; Ashmun, RA; Shapiro, LH; et al. (2000). "Aminopeptidase N is a receptor for tumor homing peptides and a target for inhibiting angiogenesis". Cancer Res. 60 (3): 722–727. PMC 4469333. PMID 10676659.
- Gridley, T (2007). "Notch signaling in vascular development and physiology". Development. 134 (15): 2709–2718. doi:10.1242/dev.004184. PMID 17611219.
- Sato, T; Tozawa, Y; Deutsch, U; Wolburg-Buchholz, K; Fujiwara, Y; Gendron-Maguire, M; Gridley, T; Wolburg, H; et al. (1995). "Distinct roles of the receptor tyrosine kinases Tie1 and Tie2 in blood vessel formation". Nature. 376 (6535): 70–74. doi:10.1038/376070a0. PMID 7596437. S2CID 4353595.
- Janes, P; Saha, N; Barton, WA; Kolev, MV; Wimmer-Kleikamp, SH; Nievergall, E; Blobel, CP; Himanen, JP; et al. (2005). "Distinct roles of the receptor tyrosine kinases Tie1 and Tie2 in blood vessel formation". Cell. 123 (2): 291–304. doi:10.1016/j.cell.2005.08.014. PMID 16239146.
- Kumar, P; Amin, MA; Harlow, LA; Polverini, PJ; Koch, AE (2003). "Src and phosphatidylinositol 3-kinase mediate soluble E-selectin induced angiogenesis". Blood. 101 (10): 3960–3968. doi:10.1182/blood-2002-04-1237. PMID 12522014.
- Roman, M; Sironi, M; Toniatti, C; Polentarutti, N; Fruscella, P; Ghezzi, P; Faggioni, R; Luini, W; et al. (1997). "Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment". Immunity. 6 (3): 315–325. doi:10.1016/S1074-7613(00)80334-9. PMID 9075932.
- Basil, J; Holmbeck, K; Bugge, TH; Gutkind, JS (2007). "MT1-MMP controls tumor induced angiogenesis through the release of semaphoring 4D". J. Biol. Chem. 282 (9): 6899–6905. doi:10.1074/jbc.M609570200. PMID 17204469.
- Lakka, S; Gondi, CS; Dinh, DH; Olivero, WC; Gujrati, M; Rao, VH; Sioka, C; Rao, JS (2005). "Specific interference of urokinase type plasminogen activatior receptor and matrix metalloproteinase 9 gene expression induced by double stranded RNA results in decreased invasion, tumor growth, and angiogenesis in gliomas". J. Biol. Chem. 280 (23): 21882–21892. doi:10.1074/jbc.M408520200. PMID 15824107.
- Dvorak, H; Brown, LF; Detmar, M; Dvorak, AM (1995). "Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis". Am. J. Pathol. 146 (5): 1029–1039. PMC 1869291. PMID 7538264.
- Bugge, T; Kombrinck, KW; Xiao, Q; Holmbäck, K; Daugherty, CC; Witte, DP; Degen, JL (1997). "Growth and dissemination of lewis lung carcinoma in plasminogen-deficient mice". Blood. 90 (11): 4522–4531. doi:10.1182/blood.V90.11.4522. PMID 9373263.
- Hiller, O; Allen, E; Apel, IJ; Gyetko, MR; Weiss, SJ (1998). "Matrix metalloproteinases regulate meovascularization by acting as pericellular fibrinolysins". Cell. 95 (3): 365–377. doi:10.1016/S0092-8674(00)81768-7. PMID 9814707.
- Kato, T; Kure, T; Chang, JH; Gabison, EE; Itoh, T; Itohara, S; Azar, DT (2001). "Diminished corneal angiogenesis in gelatinase A deficient mice". FEBS Lett. 508 (2): 187–190. doi:10.1016/S0014-5793(01)02897-6. PMID 11718713.
- Shichiri, M; Hirata, Y (2001). "Anti-angiogenesis signals by endostatin". FASEB J. 15 (6): 1044–1053. doi:10.1096/fj.99-1083com. PMID 11292666. S2CID 18206470.
- Mongiat, M (2003). "Endorepellin, a novel inhibitor of angiogenesis derived from the carboxyl terminus of perlecan". J. Biol. Chem. 278 (6): 4238–4239. doi:10.1074/jbc.M210445200. PMID 12435733.
- Abounader, R; Laterra, J (2005). "Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis". Neuro-Oncol. 7 (4): 436–451. doi:10.1215/S1152851705000050. PMC 1871724. PMID 16212809.
- Yee, J; Yan, L; Dominguez, JC; Allan, EH; Martin, TJ (1993). "Plasminogen dependent activation of latent transforming growth factor beta by growing cultures of osteoblast like cells". J. Cell. Physiol. 157 (3): 528–534. doi:10.1002/jcp.1041570312. PMID 8253864. S2CID 22817811.
- Lee, S; Jilani, SM; Nikolova, GV; Carpizo, D; Iruela-Arispe, ML (2005). "Processing of VEGF-A by marix metalloproteinases regulates bioavailaibility and vascular patterning in tumors". J. Cell Biol. 169 (4): 681–691. doi:10.1083/jcb.200409115. PMC 2171712. PMID 15911882.
- De La Luz Sierra, M; Yang, F; Narazaki, M; Salvucci, O; Davis, D; Yarchoan, R; Zhang, HH; Fales, H; Tosato, G (2004). "Differential processing of stromal-derived factor-1alpha and stromal-derived factor-1beta explains functional diversity". Blood. 103 (7): 2452–2459. doi:10.1182/blood-2003-08-2857. PMID 14525775.
- Black, R (2002). "Tumor necrosis factor-alpha converting enzyme". Int. J. Biochem. Cell Biol. 34 (1): 1–5. doi:10.1016/S1357-2725(01)00097-8. PMID 11733179.
- Brooks, P; Silletti, S; Von Schalscha, TL; Friedlander, M; Cheresh, DA (1998). "Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity". Cell. 92 (3): 391–400. doi:10.1016/S0092-8674(00)80931-9. PMID 9476898.
- Trochan-Joseph, V; Martel-Renoir, D; Mir, LM; Thomaïdis, A; Opolon, P; Connault, E; Li, H; Grenet, C; et al. (2004). "Evidence of antiangiogenic and antimetastatic activities of recombinant disintegrin domain of metargidin". Cancer Res. 64 (6): 2062–2069. doi:10.1158/0008-5472.CAN-03-3272. PMID 15026344.
- Vazquez, F; Hastings, G; Ortega, MA; Lane, TF; Oikemus, S; Lombardo, M; Iruela-Arispe, ML (1999). "METH-1 a human ortholog of ADAMTS-1 and METH-2 are members of a new family of proteins with angio-inhibitory activity". J. Biol. Chem. 274 (33): 23349–23357. doi:10.1074/jbc.274.33.23349. PMID 10438512.
- Blasi, F; Carmeliet, P (2002). "uPAR: a versatile signaling orchestrator". Nat. Rev. Mol. Cell Biol. 3 (12): 932–943. doi:10.1038/nrm977. PMID 12461559. S2CID 31211771.
- Chapman, H; Wei, Y (2001). "Protease crosstalk with integrins: the urokinase receptor paradigm". Thromb. Haemost. 86 (1): 124–129. doi:10.1055/s-0037-1616208. PMID 11486997.
- Bajou, K; Noël, A; Gerard, RD; Masson, V; Brunner, N; Holst-Hansen, C; Skobe, M; Fusenig, NE; et al. (1998). "Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization". Nat. Med. 4 (8): 923–928. doi:10.1038/nm0898-923. PMID 9701244. S2CID 36918177.
- Bajou, K; Masson, V; Gerard, RD; Schmitt, PM; Albert, V; Praus, M; Lund, LR; Frandsen, TL; et al. (2001). "The plasminogen activator inhibitor PAI-1 controls in vivo tumor vascularization by interaction with proteases, not vitronectin". J. Cell Biol. 152 (4): 777–784. doi:10.1083/jcb.152.4.777. PMC 2195770. PMID 11266468.
- Oh, J; Takahashi, R; Kondo, S; Mizoguchi, A; Adachi, E; Sasahara, RM; Nishimura, S; Imamura, Y; et al. (2001). "The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis". Cell. 107 (6): 789–800. doi:10.1016/S0092-8674(01)00597-9. hdl:2433/149674. PMID 11747814. S2CID 17290828.
- Polverini, P (1997). "Role of the macrophage in angiogenesis-dependent diseases". Regulation of Angiogenesis. pp. 11–28. doi:10.1007/978-3-0348-9006-9_2. ISBN 978-3-0348-9864-5. PMID 9002218.
{{cite book}}:|journal=ignored (help) - Rafii, S; Lyden, D (2003). "Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration". Nat. Med. 9 (6): 702–712. doi:10.1038/nm0603-702. PMID 12778169. S2CID 10294635.
- Hill, J; Kosiol, S; Schiegl, T; Ahlers, P; Walenta, K; Link, A; Böhm, M; Nickenig, G (2003). "Circulation endothelial progenitor cells, vascular function, and cardiovascular risk". N. Engl. J. Med. 353 (10): 999–1007. doi:10.1056/NEJMoa043814. PMID 16148285.
- Heissig, B; Rafii, S; Akiyama, H; Ohki, Y; Sato, Y; Rafael, T; Zhu, Z; Hicklin, DJ; et al. (2005). "Low dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9 mediated progenitor cell mobilization". J. Exp. Med. 202 (6): 739–750. doi:10.1084/jem.20050959. PMC 2212942. PMID 16157686.
- Urbich, C; Heeschen, C; Aicher, A; Sasaki, K; Bruhl, T; Farhadi, MR; Vajkoczy, P; Hofmann, WK; et al. (2005). "Cathepsin L1 is required for endothelial progenitor cell induced neovascularization". Nat. Med. 11 (2): 206–213. doi:10.1038/nm1182. PMID 15665831. S2CID 20806219.
- Nielsen, B; Sehested, M; Kjeldsen, L; Borregaard, N; Rygaard, J; Danø, K (1997). "Expression of matrix metalloproteases-9 in vascular pericytes in human breast cancer". Lab. Invest. 77 (4): 345–355. PMID 9354769.
- Chantrain, C; Shimada, H; Jodele, S; Groshen, S; Ye, W; Shalinsky, DR; Werb, Z; Coussens, LM; Declerck, YA (2004). "Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment". Cancer Res. 64 (5): 1675–1686. doi:10.1158/0008-5472.CAN-03-0160. PMID 14996727.
- Schlingemann, R; Oosterwijk, E; Wesseling, P; Rietveld, FJ; Ruiter, DJ (1996). "Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment". J. Pathol. 179 (4): 436–442. doi:10.1002/(SICI)1096-9896(199608)179:4<436::AID-PATH611>3.0.CO;2-A. hdl:2066/22707. PMID 8869294.
- Saunders, W; Bohnsack, BL; Faske, JB; Anthis, NJ; Bayless, KJ; Hirschi, KK; Davis, GE (2006). "Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3". J. Cell Biol. 175 (1): 179–191. doi:10.1083/jcb.200603176. PMC 2064509. PMID 17030988.
- Bergers, G; Song, S; Meyer-Morse, N; Bergsland, E; Hanahan, D (2003). "Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors". J. Clin. Invest. 111 (9): 1287–1295. doi:10.1172/JCI17929. PMC 154450. PMID 12727920.
- Coussens, L; Fingleton, B; Matrisian, LM (2002). "Matrix metalloproteinase inhibitors and cancer: trials and tribulations". Science. 295 (5564): 2387–2392. doi:10.1126/science.1067100. PMID 11923519. S2CID 19944201.