DNA annotation
In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome,[2] by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate.[3] Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.[4]

Annotation is performed after a genome is sequenced and assembled, and is a necessary step in genome analysis before the sequence is deposited in a database and described in a published article. Although describing individual genes and their products or functions is sufficient to consider this description as an annotation, the depth of analysis reported in literature for different genomes vary widely, with some reports including additional information that goes beyond a simple annotation.[5] Furthermore, due to the size and complexity of sequenced genomes, DNA annotation is not performed manually, but is instead automated by computational means. However, the conclusions drawn from the obtained results require manual expert analysis.[6]
DNA annotation is classified into two categories: structural annotation, which identifies and demarcates elements in a genome, and functional annotation, which assigns functions to these elements.[7] This is not the only way in which it has been categorized, as several alternatives, such as dimension-based[8] and level-based classifications,[3] have also been proposed.
History
The first generation of genome annotators used local ab initio methods, which are based solely on the information that can be extracted from the DNA sequence on a local scale, that is, one open reading frame (ORF) at a time.[9][10] They appeared as a necessity to handle the enormous amount of data produced by the Maxam-Gilbert and Sanger DNA sequencing techniques developed in the late 1970s. The first software used to analyze sequencing reads is the Staden Package, created by Rodger Staden in 1977.[11] It performed several tasks related to annotation, such as base and codon counts. In fact, codon usage was the main strategy used by several early protein coding sequence (CDS) prediction methods,[12][13][14] based on the assumption that the most translated regions in a genome contain codons with the most abundant corresponding tRNAs (the molecules responsible for carrying amino acids to the ribosome during protein synthesis) allowing a more efficient translation.[15] This was also known to be the case for synonymous codons, which are often present in proteins expressed at a lower level.[13][16]
The advent of complete genomes in the 1990s (the first one being the genome of Haemophilus influenzae sequenced in 1995) introduced a second generation of annotators. Just like in the previous generation, they performed annotation through ab initio methods, but now applied on a genome-wide scale.[9][10] Markov models are the driving force behind many algorithms used within annotators of this generation;[17][18] these models can be thought of as directed graphs where nodes represent different genomic signals (such as transcription and translation start sites) connected by arrows representing the scanning of the sequence. To ensure a Markov model detects a genomic signal, it must first be trained on a series of known genomic signals.[19] The output of Markov models in the context of annotation includes the probabilities of every kind of genomic element in every single part of the genome, and an accurate Markov model will assign high probabilities to correct annotations and low probabilities to the incorrect ones.[20]
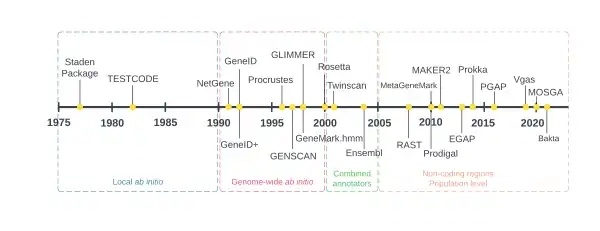
As more sequenced genomes began to be available in early and mid 2000s, coupled with the numerous protein sequences that were obtained experimentally, genome annotators began employing homology based methods, launching the third generation of genome annotation. These new methods allowed annotators not only to infer genomic elements through statistical means (as in previous generations) but could also perform their task by comparing the sequence being annotated with other already existing and validated sequences. These so called combiner annotators, which perform both ab initio and homology-based annotation, require fast alignment algorithms to identify regions of homology.[2][9][10]
In the late 2000s, genome annotation shifted its attention towards identifying non-coding regions in DNA, which was achieved thanks to the appearance of methods to analyze transcription factor binding sites, DNA methylation sites, chromatin structure, and other RNA and regulatory region analysis techniques. Other genome annotators also began to focus on population-level studies represented by the pangenome; by doing so, for instance, annotation pipelines ensure that core genes of a clade are also found in new genomes of the same clade. Both annotation strategies constitute the fourth generation of genome annotators.[9][10]
By the 2010s, the genome sequences of more than a thousand human individuals (through the 1000 Genomes Project) and several model organisms became available. As such, genome annotation remains a major challenge for scientists investigating the human and other genomes.[21][22]
Structural annotation
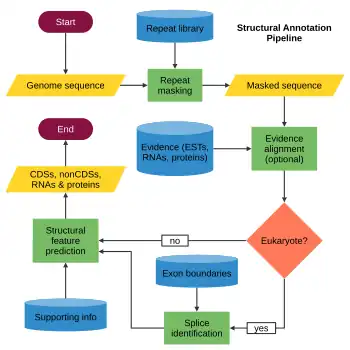
Structural annotation describes the precise location of the different elements in a genome, such as open reading frames (ORFs), coding sequences (CDS), exons, introns, repeats, splice sites, regulatory motifs, start and stop codons, and promoters.[6][23] The main steps of structural annotation are:
- Repeat identification and masking.
- Evidence alignment (optional).
- Splice identification (only in eukaryotes).
- Feature prediction (coding and noncoding sequences).
Repeat identification and masking
The first step of structural annotation consists in the identification and masking of repeats, which include low-complexity sequences (such as AGAGAGAG, or monopolymeric segments like TTTTTTTTT), and transposons (which are larger elements with several copies across the genome).[2][24] Repeats are a major component of both prokaryotic and eukaryotic genomes; for instance, between 0% and over 42% of prokaryotic genomes consist of repeats[25] and three quarters of the human genome are composed of repetitive elements.[26]
Identifying repeats is difficult for two main reasons: they are poorly conserved, and their boundaries are not clearly-defined. Because of this, repeat libraries must be built for the genome of interest, which can be accomplished with one of the following methods:[24][27]
- De novo methods. Repeats are identified by detecting and grouping pairs of sequences at different locations whose similarity is above a minimum threshold of sequence conservation in a self-genome comparison, thus requiring no prior information about repeat structure or sequences. The disadvantage of these methods is that they can identify any repeated sequence, not just transposons, and may include conserved coding sequences (CDS), making careful post-processing an indispensable step to remove these sequences. It may also leave out related regions that have degraded over time and may group elements that have no connection in their evolutionary history.[28]
- Homology-based methods. Repeats are identified by similarity (homology) of known repeats stored in a curated database. These methods are more likely to find real transposons, even in lower quantities, when compared with de novo methods, but are biased towards previously identified families.
- Structure-based methods. Repeats are identified based on models of their structure, rather than repetition or similarity. They are capable of identifying real transposons (just like the homology-based ones), but are not biased by known elements. However, they are highly specific to each class of repeat, and, as such, are less universally applicable.
- Comparative genomic methods. Repeats are identified as disruptions of one or more sequences in a multiple sequence alignment produced by large insertion regions. Although this strategy avoids the poorly-defined boundary problem that exists in other methods, it is highly dependent on assembly quality and the level of activity of transposons in the genomes in question.
After the repetitive regions in a genome have been identified, they are masked. Masking means replacing the letters of the nucleotides (A, C, G, or T) with other letters. By doing so, these regions will be marked as repetitive and downstream analyses will treat them accordingly. Repetitive regions may produce performance issues if they are not masked, and may even produce false evidence for gene annotation (for example, treating an open reading frame (ORF) in a transposon as an exon)[24] Depending on the letters used for replacement, masking can be classified as soft or hard: in soft masking, repetitive regions are indicated with lowercase letters (a, c, g, or t), whereas in hard masking, the letters of these regions are replaced with N's. This way, for example, soft masking can be used to exclude word matches and avoid initiating an alignment in those regions, and hard masking, apart from all of this, can also exclude masked regions from alignment scores.[29][30]
Evidence alignment
The next step after genome masking usually involves aligning all available transcript and protein evidence with the analyzed genome, that is, aligning all known expressed sequence tags (ESTs), RNAs and proteins of the organism being annotated with the genome.[31] Although it is optional, it can improve gene sequence elucidation because RNAs and proteins are direct products of coding sequences.[19]
If RNA-Seq data is available, it may be used to annotate and quantify all of the genes and their isoforms located in the corresponding genome, providing not only their locations, but also their rates of expression.[32] However, transcripts provide insufficient information for gene prediction because they might be unobtainable from some genes, they may encode operons of more than one gene, and their start and stop codons cannot be determined due to frameshifts and translation initiation factors.[19] To solve this problem, proteogenomics based approaches are employed, which utilize information from expressed proteins often derived from mass spectrometry.[33]
Splice identification
Annotation of eukaryotic genomes has an extra layer of difficulty due to RNA splicing, a post-transcriptional process in which introns (non-coding regions) are removed and exons (coding regions) are joined.[23] Therefore, eukaryotic coding sequences (CDS) are discontinuous, and, to ensure their proper identification, intronic regions must be filtered. To do so, annotation pipelines must find the exon-intron boundaries, and multiple methodologies have been developed for this purpose. One solution is to use known exon boundaries for alignment; for instance, many introns begin with GT and end with AG.[31] This approach, however, cannot detect novel boundaries, so alternatives like machine learning algorithms exist that are trained on known exon boundaries and quality information to predict new ones.[34] Predictors of new exon boundaries usually require efficient data-compression and alignment algorithms, but they are prone to failure in boundaries located in regions with low sequence coverage or high error-rates produced during sequencing.[35][36]
Feature prediction
A genome is divided in coding and noncoding regions, and the last step of structural annotation consists in identifying these features within the genome. In fact, the primary task in genome annotation is gene prediction, which is why numerous methods have been developed for this purpose.[19] Gene prediction is a misleading term, as most gene predictors only identify coding sequences (CDS) and do not report untranslated regions (UTRs); for this reason, CDS prediction has been proposed as a more accurate term.[24] CDS predictors detect genome features through methods called sensors, which include signal sensors that identify functional site signals such as promoters and polyA sites, and content sensors that classify DNA sequences into coding and noncoding content.[37] Whereas prokaryotic CDS predictors mostly deal with open reading frames (ORFs), which are segments of DNA between the start and stop codons, eukaryotic CDS predictors are faced with a more difficult problem because of the complex organization of eukaryotic genes.[3] CDS prediction methods can be classified into three broad categories:[2][31]
- Ab initio methods (also called statistical, intrinsic, or de novo). CDS prediction is based solely on the information that can be extracted from the DNA sequence. They rely on statistical methods such as the hidden Markov model (HMM). Some methods employ two or more genomes to infer local mutation rates and patterns along the genome.[38]
- Homology-based methods (also called empirical, evidence-driven, or extrinsic). CDS prediction is based on similarity to known sequences. Specifically, it performs alignments of the analyzed sequence with expressed sequence tags (ESTs), complementary DNA (cDNA), or protein sequences.
- Combiners. CDS prediction is done by a combination of both methods mentioned above.
Functional annotation
Functional annotation assigns functions to the genomic elements found by structural annotation,[7] by relating them to biological processes such as the cell cycle, cell death, development, metabolism, etc.[3] It may also be used as an additional quality check by identifying elements that may have been annotated by error.[2]
Coding sequence function prediction
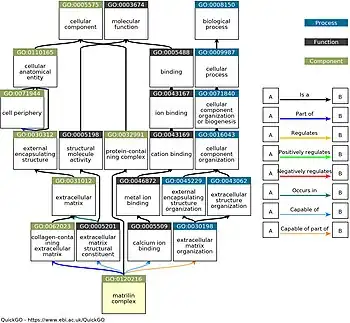
Functional annotation of genes requires a controlled vocabulary (or ontology) to name the predicted functional features. However, because there are numerous ways to define gene functions, the annotation process may be hindered when it is performed by different research groups. As such, a standardized controlled vocabulary must be employed, the most comprehensive of which is the Gene Ontology (GO). It classifies functional properties into one of three categories (molecular function, biological process, and cellular component) and organizes them in a directed acyclic graph, in which every node is a particular function, and every edge (or arrow) between two nodes indicates a parent-child or subcategory-category relationship.[40][41] As of 2020, GO is the most widely used controlled vocabulary for functional annotation of genes, followed by the MIPS Functional Catalog (FunCat).[42]
Some conventional methods for functional annotation are homology-based, which rely on local alignment search tools.[40] Its premise is that high sequence conservation between two genomic elements implies that their function is conserved as well. Pairs of homologous sequences that appeared through paralogy, orthology or orthology usually perform a similar function. However, orthologous sequences should be treated with caution because of two reasons: (1) they might have different names depending on when they were originally annotated, and (2) they may not perform the same functional role in two different organisms. Annotators often refer to an analogous sequence when no paralogy, orthology or xenology was found.[19] Homology-based methods have several drawbacks, such as errors in the database, low sensitivity/specificity, inability to distinguish between paralogy and homology,[43] artificially high scores due to the presence of low complexity regions, and significant variation within a protein family.[44]
Functional annotation can be performed through probabilistic methods. The distribution of hydrophilic and hydrophobic amino acids indicates whether a protein is located in a solution or membrane. Specific sequence motifs provide information on posttranslational modifications and final location of any given protein.[19] Probabilistic methods may be paired with a controlled vocabulary, such as GO; for example, protein-protein interaction (PPI) networks usually place proteins with similar functions close to each other.[45]
Machine learning methods are also used to generate functional annotations for novel proteins based on GO terms. Generally, they consist in constructing a binary classifier for each GO term, which are then joined together to make predictions on individual GO terms (forming a multiclass classifier) for which confidence scores are later obtained. The support vector machine (SVM) is the most widely used binary classifier in functional annotation; however, other algorithms, such as k-nearest neighbors (kNN) and convolutional neural network (CNN), have also been employed.[40]
Binary or multiclass classification methods for functional annotation generally produce less accurate results because they do not take into account the interrelations between GO terms. More advanced methods that consider these interrelations do so by either a flat or hierarchical approach, which are distinguished by the fact that the former does not take into account the ontology structure, while the latter does. Some of these methods compress the GO terms by matrix factorization or by hashing, thus boosting their performance.[42]
Noncoding sequence function prediction
Noncoding sequences (ncDNA) are those that do not code for proteins. They include elements such as pseudogenes, segmental duplications, binding sites and RNA genes.[28]
Pseudogenes are mutated copies of protein-coding genes that lost their coding function due to a disruption in their open reading frame (ORF), making them untranslatable.[28] They may be identified using one of the following two methods:[46]
- Homology-based method. Pseudogenes are identified by searching sequences that are similar to functional genes but contain mutations that produce a disruption in their ORF. This method cannot determine the evolutionary relationship between a pseudogene and its parent gene nor the elapsed time since the event happened.
- Phylogeny-based method. Pseudogenes are identified by means of a phylogenetic analysis. First, a species tree of the species of interest and a phylogenetic tree of the gene (or gene family) of interest are constructed. The two are then compared to identify a species that has lost the gene. Next, within the genome of the species where the gene was not found, a sequence is searched that is orthologous to the gene identified in the closest species. Finally, if this orthologous sequence has a disruption in its ORF (and it meets with other criteria, such as RNA-Seq data analysis, dN/dS ratio, etc.), it means that the sequence is indeed a pseudogene.
Segmental duplications are DNA segments of more than 1000 base pairs that are repeated in the genome with more than 90% sequence identity. Two strategies used for their identification are WGAC and WSSD:[47]
- Whole-Genome Assembly Comparison (WGAC). It aligns the entire genome to itself in order to identify repeated sequences after filtering out common repeats; it does not require having the original reads used for the assembly.
- Whole-genome Shotgun Sequence Detection (WSSD). It aligns the original reads with the assembled genome and searches for regions with a higher read depth than the average, which usually are signals of duplication. Segmental duplications identified by this method but not by WGAC are likely collapsed duplications, which means that they were mistakenly aligned to the same region.[48]
DNA binding sites are regions in the genome sequence that bind to and interact with specific proteins. They play an important role in DNA replication and repair, transcriptional regulation, and viral infection. Binding site prediction involves the use of one of the following two methods:[49]
- Sequence similarity based methods. They consist in the identification of homologous sequences with known DNA binding sites, or by aligning them with query proteins. Their performance is usually low because the DNA binding sequences are less conserved.
- Structure based methods. They employ the three-dimensional structural information of proteins to predict the locations of DNA binding sites.
Noncoding RNA (ncRNA), produced by RNA genes, is a type of RNA that is not translated into a protein. It includes molecules such as tRNA, rRNA, snoRNA, and microRNA, as well as noncoding mRNA-like transcripts. Ab initio prediction of RNA genes in a single genome often yields inaccurate results (with an exception being miRNA), so multi-genome comparative methods are used instead. These methods are specifically concerned with the secondary structures of ncRNA, as they are conserved in related species even when their sequence is not. Therefore, by performing a multiple sequence alignment, more useful information can be obtained for their prediction. Homology search may also be employed to identify RNA genes, but this procedure is complicated, especially in eukaryotes, due to presence of a large number of repeats and pseudogenes.[50]
Visualization

File formats
Visualization of annotations in a genome browser requires a descriptive output file, which should describe the intron-exon structures of each annotation, their start and stop codons, UTRs and alternative transcripts, and ideally should include information about the sequence alignments an gene predictions that support each gene model. Some commonly used formats for describing annotations are GenBank, GFF3, GTF, BED and EMBL.[24] Some of these formats use controlled vocabularies and ontologies to define their descriptive terminologies and guarantee interoperability between analysis and visualization tools.[2]
Genome browsers
Genomic browsers are software products that simplify the analysis and visualization of large genomic sequence and annotation data to gain biological insight, via a graphical interface. [52] [31] [53]
Genomic browsers can be divided into web-based genomic browsers and stand-alone genomic browsers. The former use information from databases and can be classified into multiple-species (integrate sequence and annotations of multiple organisms and promote cross-species comparative analysis) and species-specific (focus on one organism and the annotations for particular species). The latter are not necessarily linked to a specific genome database but are general-purpose browsers that can be downloaded and installed as an application on a local computer.[54] [19]
Comparative visualization of genomes

Comparative genomics aims to identify similarities and differences in genomic features, as well as to examine evolutionary relationships between organisms.[55] Visualization tools capable of illustrating the comparative behavior between two or more genomes are essential for this approach, and can be classified into three categories based on the representation of the relationships between the compared genomes:[19]
- Dot Plots: This scheme only allows to show the alignment of two genomes, one genome is represented along the horizontal axis and the other along the vertical axis and the dots in the plot represent the genomic elements that are similar between these two annotations.
- Linear representation: This representation uses multiple linear tracks to represent multiple genomes and their features where "track" is a concept that refers to a specific type of genomic feature at a genomic location.
- Circular representation: This representation facilitates comparison of whole microbial or viral genomes. In this visualization mode, concentric circles and arcs are used to represent genomic sections.
Quality control
The quality of the sequence assembly influences the quality of the annotation, so it is important to assess assembly quality before performing the subsequent annotation steps.[31] In order to quantify the quality of a genome annotation, three metrics have been used: recall, precision and accuracy; although these measures are not explicitly used in annotation projects, but rather in discussions of prediction accuracy.[56]
Community annotation approaches are great techniques for quality control and standardization in genome annotation. An annotation jamboree that took part in 2002, led to the creation of the annotation standards used by the Sanger Institute's Human and Vertebrate Analysis Project (HAVANA)[57].[20]
Re-annotation
Annotation projects often rely on previous annotations of an organism's genome; however, these older annotations may contain errors that can propagate to new annotations. As new genome analysis technologies are developed and richer databases become available, the annotation of some older genomes may be updated. This process, known as reannotation, can provide users with new information about the genome, including details about genes and protein functions. Re-annotation is therefore a useful approach in quality control. [56] [58]
Community annotation
Community annotation consists in the engagement of a community (both scientific and nonscientific) in genome annotation projects. It can be classified into the following six categories: [59][3]
- Factory model: Annotation is performed by a completely automated pipeline.
- Museum model: Manual curation by experts is involved to interpret the results of an annotation project.
- Cottage industry model: Annotation is decentralized and is the result of the effort from different part-time curators.
- Party or jamboree model: Consists of a short intensive workshop with leading curators from the community. It was first used in the Drosophila melanogaster genome annotation project. [60]
- Blessed annotator: A variation of the museum model, applied in the Knockout Mouse Project (KOMP), in which curators go through a training period prior to annotation, and are then given access to annotation tools to continue their work.
- Gatekeeper approach: It is a combination of the jamboree and cottage industry models. It begins with an annotation workshop, followed by a decentralized collaboration to extend and refine the initial annotation. It has been used for multiple species data.
A community annotation is said to be supervised when there is a coordinator who manages the project by requesting the annotation of specific items to a select number of experts. On the other hand, when anyone can enter a project and coordination is accomplished in a decentralized manner, it is called unsupervised community annotation. Supervised community annotation is short-lived and limited to the duration of the event, whereas the unsupervised counterpart does not have this limitation. However, the latter has been less successful than the former presumably due to a lack of time, motivation, incentive and/or communication. [61]
Wikipedia has multiple WikiProjects aimed at improving annotation. The Gene WikiProject, for instance, operates a bot that harvests gene data from research databases and creates gene stubs on that basis. [62] The RNA WikiProject seeks to write articles that describe individual RNAs and RNA families in an accessible way. [63]
Applications
Disease diagnosis
Gene Ontology is being used by researchers to establish a disease-gene relationship, as GO helps in the identification of novel genes, the alterations in their expression, distribution and function under a different set of conditions, such as diseased versus healthy.[41] Databases of this disease-gene relationships of different organisms have been created, such as Plant-Pathogen Ontology,[64] Plant-Associated Microbe Gene Ontology[65] or DisGeNET.[66] And some others have been implemented in pre-existing databases like Rat Disease Ontology in the Rat Genome database.[67]
Bioremediation
A great diversity of catabolic enzymes involved in hydrocarbon degradation by some bacterial strains are encoded by genes located in their mobile genetic elements (MGEs). The study of these elements is of great importance in the field of bioremediation, since recently the inoculation of wild or genetically modified strains with these MGEs has been sought in order to acquire these hydrocarbon degradation capacities.[68] In 2013, Phale et al.[69] published the genome annotation of a strain of Pseudomonas putida (CSV86), a bacterium known for its preference of naphthalene and other aromatic compounds over glucose as a carbon and energy source. In order to find the MGEs of this bacterium, its genome was annotated using RAST and the NCBI Prokaryotic Genome Annotation Pipeline (PGAP), and the identification of nine mobile elements was possible with the Insertion Sequence (IS) Finder database. This analysis concluded in the localization of the upper pathway genes of naphthalene degradation,[70] right next to the genes encoding tRNA-Gly and integrase, as well as the identification of the genes encoding enzymes involved in the degradation of salicylate, benzoate, 4-hydroxybenzoate, phenylacetic acid, hydroxyphenyl acetic acid, and the recognition of an operon involved in glucose transport in the strain.
Gene Ontology analysis is of great importance in functional annotation, and specifically in bioremediation it can be applied to know the relationships between the genes of some microorganisms with their functions and their role in the remediation of certain contaminants. This was the approach of the investigation and identification of Halomonas zincidurans strain B6(T), a bacterium with thirty-one genes encoding resistance to heavy metals, especially zinc[71] and Stenotrophomonas sp. DDT-1, a strain capable of using DDT as its sole carbon and energy source, [72] to mention a few examples.
Software
Genes in a eukaryotic genome can be annotated using various annotation tools[73] such as FINDER.[74] A modern annotation pipeline can support a user-friendly web interface and software containerization such as MOSGA.[75][76] Modern annotation pipelines for prokaryotic genomes are Bakta,[77] Prokka[51] and PGAP.[78]
The National Center for Biomedical Ontology develops tools for automated annotation[79] of database records based on the textual descriptions of those records.
As a general method, dcGO[80] has an automated procedure for statistically inferring associations between ontology terms and protein domains or combinations of domains from the existing gene/protein-level annotations.
A variety of software tools have been developed that allow scientists to view and share genome annotations, such as MAKER.
Genome annotation is an active area of investigation and involves a number of different organizations in the life science community which publish the results of their efforts in publicly available biological databases accessible via the web and other electronic means. Here is an alphabetical listing of on-going projects relevant to genome annotation:
References
- Zheng S, Poczai P, Hyvönen J, Tang J, Amiryousefi A (2020). "Chloroplot: An Online Program for the Versatile Plotting of Organelle Genomes". Frontiers in Genetics. 11 (576124): 576124. doi:10.3389/fgene.2020.576124. PMC 7545089. PMID 33101394.
- Dominguez Del Angel V, Hjerde E, Sterck L, Capella-Gutierrez S, Notredame C, Vinnere Pettersson O, et al. (5 February 2018). "Ten steps to get started in Genome Assembly and Annotation". F1000Research. 7 (148): 148. doi:10.12688/f1000research.13598.1. PMC 5850084. PMID 29568489.
- Stein L (July 2001). "Genome annotation: from sequence to biology". Nature Reviews. Genetics. 2 (7): 493–503. doi:10.1038/35080529. PMID 11433356. S2CID 12044602.
- Davis CP (29 March 2021). "Medical Definition of Genome annotation". MedicineNet. Archived from the original on 9 February 2023. Retrieved 17 April 2023.
- Koonin E, Galperin MY (2003). "Genome Annotation and Analysis". Sequence — Evolution — Function (1st ed.). Springer US. pp. 193–226. doi:10.1007/978-1-4757-3783-7_6. ISBN 978-1-4757-3783-7.
- Mishra P, Maurya R, Avashthi H, Mittal S, Chandra M, Ramteke PW (2021). "Genome assembly and annotation". In Singh DB, Pathak RK (eds.). Bioinformatics: Methods and Applications (1st ed.). Elsevier Science. pp. 49–66. doi:10.1016/B978-0-323-89775-4.00013-4. ISBN 9780323897754.
- Bright LA, Burgess SC, Chowdhary B, Swiderski CE, McCarthy FM (October 2009). "Structural and functional-annotation of an equine whole genome oligoarray". BMC Bioinformatics. 10 (Suppl 11): S8. doi:10.1186/1471-2105-10-S11-S8. PMC 3226197. PMID 19811692.
- Reed JL, Famili I, Thiele I, Palsson BO (February 2006). "Towards multidimensional genome annotation". Nature Reviews. Genetics. 7 (2): 130–141. doi:10.1038/nrg1769. PMID 16418748. S2CID 13107786.
- Abril JF, Castellano S (2019). "Genome Annotation". In Ranganathan S, Nakai K, Schonbach C, Gribskov M (eds.). Encyclopedia of Bioinformatics and Computational Biology (1st ed.). Elsevier Science. pp. 195–209. doi:10.1016/B978-0-12-809633-8.20226-4. ISBN 978-0-12-811432-2. S2CID 226248103.
- Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, et al. (August 2016). "NCBI prokaryotic genome annotation pipeline". Nucleic Acids Research. 44 (14): 6614–6624. doi:10.1093/nar/gkw569. PMC 5001611. PMID 27342282.
- Staden R (November 1977). "Sequence data handling by computer". Nucleic Acids Research. 4 (11): 4037–4051. doi:10.1093/nar/4.11.4037. PMC 343220. PMID 593900.
- Staden R, McLachlan AD (January 1982). "Codon preference and its use in identifying protein coding regions in long DNA sequences". Nucleic Acids Research. 10 (1): 141–156. doi:10.1093/nar/10.1.141. PMC 326122. PMID 7063399.
- Gribskov M, Devereux J, Burgess RR (January 1984). "The codon preference plot: graphic analysis of protein coding sequences and prediction of gene expression". Nucleic Acids Research. 12 (1 Pt 2): 539–549. doi:10.1093/nar/12.1part2.539. PMC 321069. PMID 6694906.
- Fickett JW (August 1996). "Finding genes by computer: the state of the art". Trends in Genetics. 12 (8): 316–320. doi:10.1016/0168-9525(96)10038-X. PMID 8783942.
- Grosjean H, Fiers W (June 1982). "Preferential codon usage in prokaryotic genes: the optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes". Gene. 18 (3): 199–209. doi:10.1016/0378-1119(82)90157-3. PMID 6751939.
- Grantham R, Gautier C, Gouy M, Mercier R, Pavé A (January 1980). "Codon catalog usage and the genome hypothesis". Nucleic Acids Research. 8 (1): r49–r62. doi:10.1093/nar/8.1.197-c. PMC 327256. PMID 6986610.
- Lukashin AV, Borodovsky M (February 1998). "GeneMark.hmm: new solutions for gene finding". Nucleic Acids Research. 26 (4): 1107–1115. doi:10.1093/nar/26.4.1107. PMC 147337. PMID 9461475.
- Salzberg SL, Delcher AL, Kasif S, White O (January 1998). "Microbial gene identification using interpolated Markov models". Nucleic Acids Research. 26 (2): 544–548. doi:10.1093/nar/26.2.544. PMC 147303. PMID 9421513.
- Soh J, Gordon PM, Sensen CW (4 September 2012). Genome Annotation. New York: Chapman and Hall/CRC. doi:10.1201/b12682. ISBN 9780429064012. Archived from the original on 18 April 2023. Retrieved 18 April 2023.
- Brent MR (December 2005). "Genome annotation past, present, and future: how to define an ORF at each locus". Genome Research. 15 (12): 1777–1786. doi:10.1101/gr.3866105. PMID 16339376.
- ENCODE Project Consortium (April 2011). Becker PB (ed.). "A user's guide to the encyclopedia of DNA elements (ENCODE)". PLOS Biology. 9 (4): e1001046. doi:10.1371/journal.pbio.1001046. PMC 3079585. PMID 21526222.

- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. (November 2012). "An integrated map of genetic variation from 1,092 human genomes". Nature. 491 (7422): 56–65. Bibcode:2012Natur.491...56T. doi:10.1038/nature11632. PMC 3498066. PMID 23128226.
- Kahl G (2015). The dictionary of genomics, transcriptomics and proteomics (Fifth ed.). Weinheim: Wiley. doi:10.1002/9783527678679. ISBN 9783527678679. Archived from the original on 4 August 2022. Retrieved 24 April 2023.
- Yandell M, Ence D (April 2012). "A beginner's guide to eukaryotic genome annotation". Nature Reviews. Genetics. 13 (5): 329–342. doi:10.1038/nrg3174. PMID 22510764. S2CID 3352427.
- Treangen TJ, Abraham AL, Touchon M, Rocha EP (May 2009). "Genesis, effects and fates of repeats in prokaryotic genomes". FEMS Microbiology Reviews. 33 (3): 539–571. doi:10.1111/j.1574-6976.2009.00169.x. PMID 19396957.
- Liehr T (February 2021). "Repetitive Elements in Humans". International Journal of Molecular Sciences. 22 (4): 2072. doi:10.3390/ijms22042072. PMC 7922087. PMID 33669810.
- Bergman CM, Quesneville H (November 2007). "Discovering and detecting transposable elements in genome sequences". Briefings in Bioinformatics. 8 (6): 382–392. doi:10.1093/bib/bbm048. PMID 17932080.
- Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB (August 2010). "Annotating non-coding regions of the genome". Nature Reviews. Genetics. 11 (8): 559–571. doi:10.1038/nrg2814. PMID 20628352. S2CID 6617359.
- Edgar RC (October 2010). "Search and clustering orders of magnitude faster than BLAST". Bioinformatics. 26 (19): 2460–2461. doi:10.1093/bioinformatics/btq461. PMID 20709691.
- Edgar R. "Sequence masking". drive5.com. Archived from the original on 3 February 2020. Retrieved 25 April 2023.
- Ejigu GF, Jung J (September 2020). "Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing". Biology. 9 (9): 295. doi:10.3390/biology9090295. PMC 7565776. PMID 32962098.
- Garber M, Grabherr MG, Guttman M, Trapnell C (June 2011). "Computational methods for transcriptome annotation and quantification using RNA-seq". Nature Methods. 8 (6): 469–477. doi:10.1038/nmeth.1613. PMID 21623353. S2CID 205419756.
- Gupta N, Tanner S, Jaitly N, Adkins JN, Lipton M, Edwards R, et al. (September 2007). "Whole proteome analysis of post-translational modifications: applications of mass-spectrometry for proteogenomic annotation". Genome Research. 17 (9): 1362–1377. doi:10.1101/gr.6427907. PMC 1950905. PMID 17690205.
- De Bona F, Ossowski S, Schneeberger K, Rätsch G (August 2008). "Optimal spliced alignments of short sequence reads". Bioinformatics. 24 (16): i174–i180. doi:10.1093/bioinformatics/btn300. PMID 18689821.
- Trapnell C, Pachter L, Salzberg SL (May 2009). "TopHat: discovering splice junctions with RNA-Seq". Bioinformatics. 25 (9): 1105–1111. doi:10.1093/bioinformatics/btp120. PMC 2672628. PMID 19289445.
- Križanovic K, Echchiki A, Roux J, Šikic M (March 2018). "Evaluation of tools for long read RNA-seq splice-aware alignment". Bioinformatics. 34 (5): 748–754. doi:10.1093/bioinformatics/btx668. PMC 6192213. PMID 29069314.
- McHardy AC, Kloetgen A (2017). "Finding Genes in Genome Sequence". In Keith JM (ed.). Bioinformatics. Methods in Molecular Biology. Vol. 1525 (Second ed.). New York: Springer. pp. 271–291. doi:10.1007/978-1-4939-6622-6_11. ISBN 978-1-4939-6622-6. PMID 27896725.
- Brent MR, Guigó R (June 2004). "Recent advances in gene structure prediction". Current Opinion in Structural Biology. 14 (3): 264–272. doi:10.1016/j.sbi.2004.05.007. PMID 15193305.
- Binns D, Dimmer E, Huntley R, Barrell D, O'Donovan C, Apweiler R (November 2009). "QuickGO: a web-based tool for Gene Ontology searching". Bioinformatics. 25 (22): 3045–3046. doi:10.1093/bioinformatics/btp536. PMC 2773257. PMID 19744993.
- Vu TT, Jung J (2021). "Protein function prediction with gene ontology: from traditional to deep learning models". PeerJ. 9: e12019. doi:10.7717/peerj.12019. PMC 8395570. PMID 34513334.
- Saxena R, Bishnoi R, Singla D (2021). "Gene Ontology: application and importance in functional annotation of the genomic data". In Singh B, Pathak RK (eds.). Bioinformatics : methods and applications. London: Academic Press. pp. 145–157. doi:10.1016/B978-0-323-89775-4.00015-8. ISBN 978-0-323-89775-4.
- Zhao Y, Wang J, Chen J, Zhang X, Guo M, Yu G (2020). "A Literature Review of Gene Function Prediction by Modeling Gene Ontology". Frontiers in Genetics. 11: 400. doi:10.3389/fgene.2020.00400. PMC 7193026. PMID 32391061.
- Sasson O, Kaplan N, Linial M (June 2006). "Functional annotation prediction: all for one and one for all". Protein Science. 15 (6): 1557–1562. doi:10.1110/ps.062185706. PMC 2242553. PMID 16672244.
- Sinha S, Lynn AM, Desai DK (October 2020). "Implementation of homology based and non-homology based computational methods for the identification and annotation of orphan enzymes: using Mycobacterium tuberculosis H37Rv as a case study". BMC Bioinformatics. 21 (1): 466. doi:10.1186/s12859-020-03794-x. PMC 574302. PMID 33076816.
- Letovsky S, Kasif S (2003). "Predicting protein function from protein/protein interaction data: a probabilistic approach". Bioinformatics. 19 (Suppl 1): i197–i204. doi:10.1093/bioinformatics/btg1026. PMID 12855458.
- Dainat J, Pontarotti P (2021). "Methods to Identify and Study the Evolution of Pseudogenes Using a Phylogenetic Approach" (PDF). In Poliseno L (ed.). Pseudogenes. Methods in Molecular Biology. Vol. 2324 (Second ed.). New York: Springer. pp. 21–34. doi:10.1007/978-1-0716-1503-4_2. ISBN 978-1-0716-1503-4. PMID 34165706. S2CID 235625288.
- Numanagic I, Gökkaya AS, Zhang L, Berger B, Alkan C, Hach F (September 2018). "Fast characterization of segmental duplications in genome assemblies". Bioinformatics. 34 (17): i706–i714. doi:10.1093/bioinformatics/bty586. PMC 6129265. PMID 30423092.
- Hartasánchez DA, Brasó-Vives M, Heredia-Genestar JM, Pybus M, Navarro A (November 2018). "Effect of Collapsed Duplications on Diversity Estimates: What to Expect". Genome Biology and Evolution. 10 (11): 2899–2905. doi:10.1093/gbe/evy223. PMC 6239678. PMID 30364947.
- Si J, Zhao R, Wu R (March 2015). "An overview of the prediction of protein DNA-binding sites". International Journal of Molecular Sciences. 16 (3): 5194–5215. doi:10.3390/ijms16035194. PMC 4394471. PMID 25756377.
- Griffiths-Jones S (2007). "Annotating noncoding RNA genes". Annual Review of Genomics and Human Genetics. 8: 279–298. doi:10.1146/annurev.genom.8.080706.092419. PMID 17506659.
- Seemann T (July 2014). "Prokka: rapid prokaryotic genome annotation". Bioinformatics. 30 (14): 2068–2069. doi:10.1093/bioinformatics/btu153. PMID 24642063.
- Valeev T, Yevshin I, Kolpakov F (2013). "BioUML Genome Browser". Virtual Biology. 1 (1): 15. doi:10.12704/vb/e8.
- Szot PS, Yang A, Wang X, Parsania C, Röhm U, Wong KH, Ho JW (May 2017). "PBrowse: a web-based platform for real-time collaborative exploration of genomic data". Nucleic Acids Research. 45 (9): e67. doi:10.1093/nar/gkw1358. PMC 5605237. PMID 28100700.
- Wang J, Kong L, Gao G, Luo J (March 2013). "A brief introduction to web-based genome browsers". Briefings in Bioinformatics. 14 (2): 131–143. doi:10.1093/bib/bbs029. PMID 22764121.
- Jung J, Kim JI, Yi G (December 2019). "geneCo: a visualized comparative genomic method to analyze multiple genome structures". Bioinformatics. 35 (24): 5303–5305. doi:10.1093/bioinformatics/btz596. PMC 6954651. PMID 31350879.
- Ouzounis CA, Karp PD (2002). "The past, present and future of genome-wide re-annotation". Genome Biology. 3 (2): COMMENT2001. doi:10.1186/gb-2002-3-2-comment2001. PMC 139008. PMID 11864365.
- "Manual Annotation - Wellcome Sanger Institute". www.sanger.ac.uk. Archived from the original on 2 February 2023. Retrieved 28 March 2023.
- Siezen RJ, van Hijum SA (July 2010). "Genome (re-)annotation and open-source annotation pipelines". Microbial Biotechnology. 3 (4): 362–369. doi:10.1111/j.1751-7915.2010.00191.x. PMC 3815804. PMID 21255336.
- Loveland JE, Gilbert JG, Griffiths E, Harrow JL (2012). "Community gene annotation in practice". Database. 2012 (2012): bas009. doi:10.1093/database/bas009. PMC 3308165. PMID 22434843.
- Hartl DL (April 2000). "Fly meets shotgun: shotgun wins". Nature Genetics. 24 (4): 327–328. doi:10.1038/74125. PMID 10742085. S2CID 5354139.
- Mazumder R, Natale DA, Julio JA, Yeh LS, Wu CH (February 2010). "Community annotation in biology". Biology Direct. 5 (1): 12. doi:10.1186/1745-6150-5-12. PMC 2834641. PMID 20167071.
- Huss JW, Orozco C, Goodale J, Wu C, Batalov S, Vickers TJ, et al. (July 2008). "A gene wiki for community annotation of gene function". PLOS Biology. 6 (7): e175. doi:10.1371/journal.pbio.0060175. PMC 2443188. PMID 18613750.
- Daub J, Gardner PP, Tate J, Ramsköld D, Manske M, Scott WG, et al. (December 2008). "The RNA WikiProject: community annotation of RNA families". RNA. 14 (12): 2462–2464. doi:10.1261/rna.1200508. PMC 2590952. PMID 18945806.
- Cooper L, Jaiswal P (2016). "The Plant Ontology: A Tool for Plant Genomics". In Edwards D (ed.). Plant Bioinformatics. Methods in Molecular Biology. Vol. 1374 (2nd ed.). Totowa, N.J.: Humana Press. pp. 89–114. doi:10.1007/978-1-4939-3167-5_5. ISBN 978-1-4939-3167-5. PMID 26519402.
- Torto-Alalibo T, Collmer CW, Gwinn-Giglio M (February 2009). "The Plant-Associated Microbe Gene Ontology (PAMGO) Consortium: community development of new Gene Ontology terms describing biological processes involved in microbe-host interactions". BMC Microbiology. 9 (Suppl 1): S1. doi:10.1186/1471-2180-9-S1-S1. PMC 2654661. PMID 19278549.
- Piñero J, Ramírez-Anguita JM, Saüch-Pitarch J, Ronzano F, Centeno E, Sanz F, Furlong LI (January 2020). "The DisGeNET knowledge platform for disease genomics: 2019 update". Nucleic Acids Research. 48 (D1): D845–D855. doi:10.1093/nar/gkz1021. PMC 7145631. PMID 31680165.
- Hayman GT, Laulederkind SJ, Smith JR, Wang SJ, Petri V, Nigam R, et al. (2016). "The Disease Portals, disease-gene annotation and the RGD disease ontology at the Rat Genome Database". Database. 2016: baw034. doi:10.1093/database/baw034. PMC 4805243. PMID 27009807.
- Top EM, Springael D, Boon N (November 2002). "Catabolic mobile genetic elements and their potential use in bioaugmentation of polluted soils and waters". FEMS Microbiology Ecology. 42 (2): 199–208. doi:10.1111/j.1574-6941.2002.tb01009.x. PMID 19709279. S2CID 15173391.
- Phale PS, Paliwal V, Raju SC, Modak A, Purohit HJ (January 2013). "Genome Sequence of Naphthalene-Degrading Soil Bacterium Pseudomonas putida CSV86". Genome Announcements. 1 (1): 234–235. doi:10.1128/genomeA.00234-12. PMC 3587945. PMID 23469351.
- Trivedi VD, Jangir PK, Sharma R, Phale PS (December 2016). "Insights into functional and evolutionary analysis of carbaryl metabolic pathway from Pseudomonas sp. strain C5pp". Scientific Reports. 6 (1): 38430. Bibcode:2016NatSR...638430T. doi:10.1038/srep38430. PMC 5141477. PMID 27924916.
- Huo YY, Li ZY, Cheng H, Wang CS, Xu XW (2014). "High quality draft genome sequence of the heavy metal resistant bacterium Halomonas zincidurans type strain B6(T)". Standards in Genomic Sciences. 9 (30): 30. doi:10.1186/1944-3277-9-30. PMC 4286145. PMID 25945155.
- Pan X, Lin D, Zheng Y, Zhang Q, Yin Y, Cai L, et al. (February 2016). "Biodegradation of DDT by Stenotrophomonas sp. DDT-1: Characterization and genome functional analysis". Scientific Reports. 6 (1): 21332. Bibcode:2016NatSR...621332P. doi:10.1038/srep21332. PMC 4758049. PMID 26888254.
- GAAS, NBIS -- National Bioinformatics Infrastructure Sweden, 13 April 2022, retrieved 25 April 2022
- Banerjee S, Bhandary P, Woodhouse M, Sen TZ, Wise RP, Andorf CM (April 2021). "FINDER: an automated software package to annotate eukaryotic genes from RNA-Seq data and associated protein sequences". BMC Bioinformatics. 22 (1): 205. doi:10.1186/s12859-021-04120-9. PMC 8056616. PMID 33879057.
- Martin R, Hackl T, Hattab G, Fischer MG, Heider D (April 2021). Birol I (ed.). "MOSGA: Modular Open-Source Genome Annotator". Bioinformatics. 36 (22–23): 5514–5515. doi:10.1093/bioinformatics/btaa1003. hdl:21.11116/0000-0006-FED4-D. PMID 33258916.
- Martin R. "MOSGA". mosga.mathematik.uni-marburg.de. Retrieved 25 April 2022.
- Schwengers O, Jelonek L, Dieckmann MA, Beyvers S, Blom J, Goesmann A (November 2021). "Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification". Microbial Genomics. 7 (11). doi:10.1099/mgen.0.000685. PMC 8743544. PMID 34739369.
- Li W, O'Neill KR, Haft DH, DiCuccio M, Chetvernin V, Badretdin A, et al. (January 2021). "RefSeq: expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation". Nucleic Acids Research. 49 (D1): D1020–D1028. doi:10.1093/nar/gkaa1105. PMC 7779008. PMID 33270901.
- "NCBO Annotator". ncbo.bioontology.org. Retrieved 8 February 2023.
- Fang H, Gough J (January 2013). "DcGO: database of domain-centric ontologies on functions, phenotypes, diseases and more". Nucleic Acids Research. 41 (Database issue): D536–D544. doi:10.1093/nar/gks1080. PMC 3531119. PMID 23161684.