Biocatalysis
Biocatalysis refers to the use of living (biological) systems or their parts to speed up (catalyze) chemical reactions. In biocatalytic processes, natural catalysts, such as enzymes, perform chemical transformations on organic compounds. Both enzymes that have been more or less isolated and enzymes still residing inside living cells are employed for this task.[1][2][3] Modern biotechnology, specifically directed evolution, has made the production of modified or non-natural enzymes possible. This has enabled the development of enzymes that can catalyze novel small molecule transformations that may be difficult or impossible using classical synthetic organic chemistry. Utilizing natural or modified enzymes to perform organic synthesis is termed chemoenzymatic synthesis; the reactions performed by the enzyme are classified as chemoenzymatic reactions.
History
Biocatalysis underpins some of the oldest chemical transformations known to humans, for brewing predates recorded history. The oldest records of brewing are about 6000 years old and refer to the Sumerians.
The employment of enzymes and whole cells have been important for many industries for centuries. The most obvious uses have been in the food and drink businesses where the production of wine, beer, cheese etc. is dependent on the effects of the microorganisms.
More than one hundred years ago, biocatalysis was employed to do chemical transformations on non-natural man-made organic compounds, with the last 30 years seeing a substantial increase in the application of biocatalysis to produce fine chemicals, especially for the pharmaceutical industry.[4]
Since biocatalysis deals with enzymes and microorganisms, it is historically classified separately from "homogeneous catalysis" and "heterogeneous catalysis". However, mechanistically speaking, biocatalysis is simply a special case of heterogeneous catalysis.[5]
Advantages of chemoenzymatic synthesis
-Enzymes are environmentally benign, being completely degraded in the environment.
-Most enzymes typically function under mild or biological conditions, which minimizes problems of undesired side-reactions such as decomposition, isomerization, racemization and rearrangement, which often plague traditional methodology.
-Enzymes selected for chemoenzymatic synthesis can be immobilized on a solid support. These immobilized enzymes demonstrate improved stability and re-usability.
-Through the development of protein engineering, specifically site-directed mutagenesis and directed evolution, enzymes can be modified to enable non-natural reactivity. Modifications may also allow for a broader substrate range, enhance reaction rate or catalyst turnover.
-Enzymes exhibit extreme selectivity towards their substrates. Typically enzymes display three major types of selectivity:
- Chemoselectivity: Since the purpose of an enzyme is to act on a single type of functional group, other sensitive functionalities, which would normally react to a certain extent under chemical catalysis, survive. As a result, biocatalytic reactions tend to be "cleaner" and laborious purification of product(s) from impurities emerging through side-reactions can largely be omitted.
- Regioselectivity and diastereoselectivity: Due to their complex three-dimensional structure, enzymes may distinguish between functional groups which are chemically situated in different regions of the substrate molecule.
- Enantioselectivity: Since almost all enzymes are made from L-amino acids, enzymes are chiral catalysts. As a consequence, any type of chirality present in the substrate molecule is "recognized" upon the formation of the enzyme-substrate complex. Thus a prochiral substrate may be transformed into an optically active product and both enantiomers of a racemic substrate may react at different rates.
These reasons, and especially the latter, are the major reasons why synthetic chemists have become interested in biocatalysis. This interest in turn is mainly due to the need to synthesize enantiopure compounds as chiral building blocks for Pharmaceutical drugs and agrochemicals.
Asymmetric biocatalysis
The use of biocatalysis to obtain enantiopure compounds can be divided into two different methods:
- Kinetic resolution of a racemic mixture
- Biocatalyzed asymmetric synthesis
In kinetic resolution of a racemic mixture, the presence of a chiral object (the enzyme) converts one of the stereoisomers of the reactant into its product at a greater reaction rate than for the other reactant stereoisomer. The stereochemical mixture has now been transformed into a mixture of two different compounds, making them separable by normal methodology.
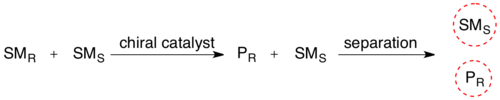
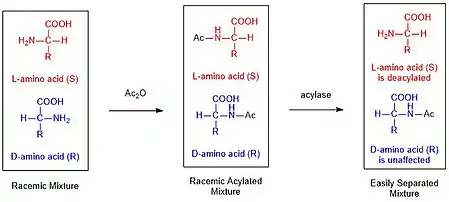
Biocatalyzed kinetic resolution is utilized extensively in the purification of racemic mixtures of synthetic amino acids. Many popular amino acid synthesis routes, such as the Strecker Synthesis, result in a mixture of R and S enantiomers. This mixture can be purified by (I) acylating the amine using an anhydride and then (II) selectively deacylating only the L enantiomer using hog kidney acylase.[6] These enzymes are typically extremely selective for one enantiomer leading to very large differences in rate, allowing for selective deacylation.[7] Finally the two products are now separable by classical techniques, such as chromatography.
The maximum yield in such kinetic resolutions is 50%, since a yield of more than 50% means that some of wrong isomer also has reacted, giving a lower enantiomeric excess. Such reactions must therefore be terminated before equilibrium is reached. If it is possible to perform such resolutions under conditions where the two substrate- enantiomers are racemizing continuously, all substrate may in theory be converted into enantiopure product. This is called dynamic resolution.
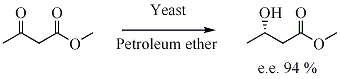
In biocatalyzed asymmetric synthesis, a non-chiral unit becomes chiral in such a way that the different possible stereoisomers are formed in different quantities. The chirality is introduced into the substrate by influence of enzyme, which is chiral. Yeast is a biocatalyst for the enantioselective reduction of ketones.
The Baeyer–Villiger oxidation is another example of a biocatalytic reaction. In one study a specially designed mutant of Candida antarctica was found to be an effective catalyst for the Michael addition of acrolein with acetylacetone at 20 °C in absence of additional solvent.[8]
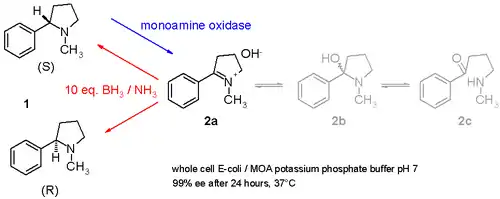
Another study demonstrates how racemic nicotine (mixture of S and R-enantiomers 1 in scheme 3) can be deracemized in a one-pot procedure involving a monoamine oxidase isolated from Aspergillus niger which is able to oxidize only the amine S-enantiomer to the imine 2 and involving an ammonia–borane reducing couple which can reduce the imine 2 back to the amine 1.[9] In this way the S-enantiomer will continuously be consumed by the enzyme while the R-enantiomer accumulates. It is even possible to stereoinvert pure S to pure R.
Photoredox enabled biocatalysis
Recently, photoredox catalysis has been applied to biocatalysis, enabling unique, previously inaccessible transformations. Photoredox chemistry relies upon light to generate free radical intermediates.[10] These radical intermediates are achiral thus racemic mixtures of product are obtained when no external chiral environment is provided. Enzymes can provide this chiral environment within the active site and stabilize a particular conformation and favoring formation of one, enantiopure product.[11] Photoredox enabled biocatalysis reactions fall into two categories:
- Internal coenzyme/cofactor photocatalyst
- External photocatalyst
Certain common hydrogen atom transfer (HAT) cofactors (NADPH and Flavin) can operate as single electron transfer (SET) reagents.[11][12][13] Although these species are capable of HAT without irradiation, their redox potentials are enhance by nearly 2.0 V upon visible light irradiation.[14] When paired with their respective enzymes (typically ene-reductases) This phenomenon has been utilized by chemists to develop enantioselective reduction methodologies. For example medium sized lactams can be synthesized in the chiral environment of an ene-reductase through a reductive, baldwin favored, radical cyclization terminated by enantioselective HAT from NADPH.[15]
The second category of photoredox enabled biocatalytic reactions use an external photocatalyst (PC). Many types of PCs with a large range of redox potentials can be utilized, allowing for greater tunability of reactive compared to using a cofactor. Rose bengal, and external PC, was utilized in tandem with an oxidoreductase to enantioselectively deacylate medium sized alpha-acyl-ketones.[16]
Using an external PC has some downsides. For example, external PCs typically complicate reaction design because the PC may react with both the bound and unbound substrate. If a reaction occurs between the unbound substrate and the PC, enantioselectivity is lost and other side reactions may occur.
Further reading
- Mortison, JD; Sherman, DH (2010). "Frontiers and opportunities in chemoenzymatic synthesis". J Org Chem. 75 (21): 7041–51. doi:10.1021/jo101124n. PMC 2966535. PMID 20882949.
- Kim, Jinhyun; Lee, Sahng Ha; Tieves, Florian; Paul, Caroline E.; Hollmann, Frank; Park, Chan Beum (5 July 2019). "Nicotinamide adenine dinucleotide as a photocatalyst". Science Advances. 5 (7): eaax0501. doi:10.1126/sciadv.aax0501.[17]
See also
References
- Anthonsen, Thorlief (2000). "Reactions Catalyzed by Enzymes". In Adlercreutz, Patrick; Straathof, Adrie J. J. (eds.). Applied Biocatalysis (2nd ed.). Taylor & Francis. pp. 18–59. ISBN 978-9058230249.
- Faber, Kurt (2011). Biotransformations in Organic Chemistry (6th ed.). Springer. ISBN 9783642173936.
- Jayasinghe, Leonard Y.; Smallridge, Andrew J.; Trewhella, Maurie A. (1993). "The yeast mediated reduction of ethyl acetoacetate in petroleum ether". Tetrahedron Letters. 34 (24): 3949–3950. doi:10.1016/S0040-4039(00)79272-0.
- Liese, Andreas; Seelbach, Karsten; Wandrey, Christian, eds. (2006). Industrial Biotransformations (2nd ed.). John Wiley & Sons. p. 556. ISBN 978-3527310012.
- Rothenberg, Gadi (2008). Catalysis: Concepts and green applications. Wiley. ISBN 9783527318247.
- Wade, L. G., 1947- (2013). Organic chemistry (8th ed.). Boston: Pearson. ISBN 978-0-321-76841-4. OCLC 752068109.
{{cite book}}: CS1 maint: multiple names: authors list (link) - Shviadas, V. Iu; Galaev, I. Iu; Galstian, N. A.; Berezin, I. V. (August 1980). "[Substrate specificity of acylase I from pig kidney]". Biokhimiia (Moscow, Russia). 45 (8): 1361–1364. ISSN 0320-9725. PMID 7236787.
- Svedendahl, Maria; Hult, Karl; Berglund, Per (December 2005). "Fast Carbon-Carbon Bond Formation by a Promiscuous Lipase". Journal of the American Chemical Society. 127 (51): 17988–17989. doi:10.1021/ja056660r. PMID 16366534.
- Dunsmore, Colin J.; Carr, Reuben; Fleming, Toni; Turner, Nicholas J. (2006). "A Chemo-Enzymatic Route to Enantiomerically Pure Cyclic Tertiary Amines". Journal of the American Chemical Society. 128 (7): 2224–2225. doi:10.1021/ja058536d. PMID 16478171.
- Prier, Christopher K.; Rankic, Danica A.; MacMillan, David W. C. (2013-07-10). "Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis". Chemical Reviews. 113 (7): 5322–5363. doi:10.1021/cr300503r. ISSN 0009-2665. PMC 4028850. PMID 23509883.
- Nakano, Yuji; Biegasiewicz, Kyle F; Hyster, Todd K (April 2019). "Biocatalytic hydrogen atom transfer: an invigorating approach to free-radical reactions". Current Opinion in Chemical Biology. 49: 16–24. doi:10.1016/j.cbpa.2018.09.001. PMC 6437003. PMID 30269010.
- Sandoval, Braddock A.; Meichan, Andrew J.; Hyster, Todd K. (2017-08-23). "Enantioselective Hydrogen Atom Transfer: Discovery of Catalytic Promiscuity in Flavin-Dependent 'Ene'-Reductases". Journal of the American Chemical Society. 139 (33): 11313–11316. doi:10.1021/jacs.7b05468. ISSN 0002-7863. PMID 28780870.
- Li, Zhining; Wang, Zexu; Meng, Ge; Lu, Hong; Huang, Zedu; Chen, Fener (April 2018). "Identification of an Ene Reductase from Yeast Kluyveromyces Marxianus and Application in the Asymmetric Synthesis of ( R )-Profen Esters". Asian Journal of Organic Chemistry. 7 (4): 763–769. doi:10.1002/ajoc.201800059.
- Emmanuel, Megan A.; Greenberg, Norman R.; Oblinsky, Daniel G.; Hyster, Todd K. (December 14, 2016). "Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light". Nature. 540 (7633): 414–417. Bibcode:2016Natur.540..414E. doi:10.1038/nature20569. ISSN 1476-4687. PMID 27974767. S2CID 205252473.
- Biegasiewicz, Kyle F.; Cooper, Simon J.; Gao, Xin; Oblinsky, Daniel G.; Kim, Ji Hye; Garfinkle, Samuel E.; Joyce, Leo A.; Sandoval, Braddock A.; Scholes, Gregory D.; Hyster, Todd K. (2019-06-21). "Photoexcitation of flavoenzymes enables a stereoselective radical cyclization". Science. 364 (6446): 1166–1169. Bibcode:2019Sci...364.1166B. doi:10.1126/science.aaw1143. ISSN 0036-8075. PMC 7028431. PMID 31221855.
- Biegasiewicz, Kyle F.; Cooper, Simon J.; Emmanuel, Megan A.; Miller, David C.; Hyster, Todd K. (July 2018). "Catalytic promiscuity enabled by photoredox catalysis in nicotinamide-dependent oxidoreductases". Nature Chemistry. 10 (7): 770–775. Bibcode:2018NatCh..10..770B. doi:10.1038/s41557-018-0059-y. ISSN 1755-4330. PMID 29892028. S2CID 48360817.
- Kim, Jinhyun; Lee, Sahng Ha; Tieves, Florian; Paul, Caroline E.; Hollmann, Frank; Park, Chan Beum (5 July 2019). "Nicotinamide adenine dinucleotide as a photocatalyst". Science Advances. 5 (7): eaax0501. Bibcode:2019SciA....5..501K. doi:10.1126/sciadv.aax0501. PMC 6641943. PMID 31334353.
External links
- Austrian Centre of Industrial Biotechnology official website
- The Centre of Excellence for Biocatalysis - CoEBio3
- The University of Exeter - Biocatalysis Centre
- Center for Biocatalysis and Bioprocessing - The University of Iowa
- TU Delft - Biocatalysis & Organic Chemistry (BOC)
- KTH Stockholm - Biocatalysis Research Group
- Institute of Technical Biocatalysis at the Hamburg University of Technology (TUHH)
- Biocascades Project