Backbone-dependent rotamer library
In biochemistry, a backbone-dependent rotamer library provides the frequencies, mean dihedral angles, and standard deviations of the discrete conformations (known as rotamers) of the amino acid side chains in proteins as a function of the backbone dihedral angles φ and ψ of the Ramachandran map. By contrast, backbone-independent rotamer libraries express the frequencies and mean dihedral angles for all side chains in proteins, regardless of the backbone conformation of each residue type. Backbone-dependent rotamer libraries have been shown to have significant advantages over backbone-independent rotamer libraries, principally when used as an energy term, by speeding up search times of side-chain packing algorithms used in protein structure prediction and protein design.[1]

History
The first backbone-dependent rotamer library was developed in 1993 by Roland Dunbrack and Martin Karplus to assist the prediction of the Cartesian coordinates of a protein's side chains given the experimentally determined or predicted Cartesian coordinates of its main chain.[2] The library was derived from the structures of 132 proteins from the Protein Data Bank with resolution of 2.0 Å or better. The library provided the counts and frequencies of χ1 or χ1+χ2 rotamers of 18 amino acids (excluding glycine and alanine residue types, since they do not have a χ1 dihedral) for each 20° x 20° bin of the Ramachandran map (φ,ψ = -180° to -160°, -160° to -140° etc.).
In 1997, Dunbrack and Fred E. Cohen at the University of California, San Francisco presented a backbone-dependent rotamer library derived from Bayesian statistics.[3] The Bayesian approach provided the opportunity for the definition of a Bayesian prior for the frequencies of rotamers in each 10° x 10° bin derived by assuming that the steric and electrostatic effects of the φ and ψ dihedral angles are independent. In addition, a periodic kernel with 180° periodicity was used to count side chains 180° away in each direction from the bin of interest. As an exponent of a sin2 function, it behaved much like a von Mises distribution commonly used in directional statistics. The 1997 library was made publicly available via the World Wide Web in 1997, and found early use in protein structure prediction[4] and protein design.[5] The library derived from Bayesian statistics was updated in 2002[6]

Many modeling programs, such as Rosetta, use a backbone-dependent rotamer library as a scoring function (usually in the form E=-ln(p(rotamer(i) | φ,ψ)) for the ith rotamer, and optimize the backbone conformation of proteins by minimizing the rotamer energy with derivatives of the log probabilities with respect to φ,ψ.[7] This requires smooth probability functions with smooth derivatives, because most mathematical optimization algorithms use first and sometimes second derivatives and will get stuck in local minima on rough surfaces. In 2011, Shapovalov and Dunbrack published a smoothed backbone-dependent rotamer library derived from kernel density estimates and kernel regressions with von Mises distribution kernels on the φ,ψ variables.[8] The treatment of the non-rotameric degrees of freedom (those dihedral angles not about sp3-sp3 bonds, such as asparagine and aspartate χ2, phenylalanine, tyrosine, histidine, tryptophan χ2, and glutamine and glutamate χ3) was improved by modeling the dihedral angle probability density of each of these dihedral angles as a function of χ1 rotamer (or χ1 and χ2 for Gln and Glu) and φ,ψ. The functions are essentially regressions of a periodic probability density on a torus.
In addition to statistical analysis of structures in the Protein Data Bank, backbone-dependent rotamer libraries can also be derived from molecular dynamics simulations of proteins, as demonstrated by the Dynameomics Library from Valerie Daggett's research group.[9] Because these libraries are based on sampling from simulations, they can generate far larger numbers of data points across regions of the Ramachandran map that are sparsely populated in experimental structures, leading to higher statistical significance in these regions. Rotamer libraries derived from simulations are dependent on the force field used in the simulations. The Dynameomics Library is built on simulations using the ENCAD force field of Levitt et al. from 1995.[10]
Backbone-dependence of rotamer populations
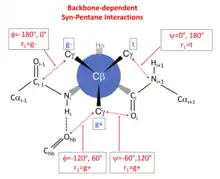
The effect of backbone conformation on side-chain rotamer frequencies is primarily due to steric repulsions between backbone atoms whose position is dependent on φ and ψ and the side-chain γ heavy atoms (carbon, oxygen, or sulfur) of each residue type (PDB atom types CG, CG1, CG2, OG, OG1, SG). These occur in predictable combinations that depend on the dihedrals connecting the backbone atoms to the side-chain atoms.[11][3] These steric interactions occur when the connecting dihedral angles form a pair of dihedral angles with values {-60°,+60°} or {+60°,-60°}, in a manner related to the phenomenon of pentane interference. For example, the nitrogen atom of residue i+1 is connected to the γ heavy atom of any side chain by a connected set of 5 atoms: N(i+1)-C(i)-Cα(i)-Cβ(i)-Cγ(i). The dihedral angle N(i+1)-C(i)-Cα(i)-Cβ(i) is equal to ψ+120°, and C(i)-Cα(i)-Cβ(i)-Cγ(i) is equal to χ1-120°. When ψ is -60° and χ1 is +60° (the g+ rotamer of a side chain), there is a steric interaction between N(i+1) and Cγ because the dihedral angles connecting them are N(i+1)-C(i)-Cα(i)-Cβ(i) = ψ+120° = +60°, and C(i)-Cα(i)-Cβ(i)-Cγ(i) = χ1-120° = -60°. The same interaction occurs when ψ is 0° and χ1 is 180° (the trans rotamer of a side chain). The carbonyl oxygen of residue i plays the same role when ψ=-60° for the g+ rotamer and when ψ=180° for the trans rotamer. Finally, φ-dependent interactions occur between the side-chain γ heavy atoms in g- and g+ rotamers on the one hand, and the carbonyl carbon of residue i-1 and a γ heavy atom, and between the backbone NH of residue i and its hydrogen-bonding partner on the other.

The φ,ψ-dependent interactions of backbone atoms and side-chain Cγ atoms can be observed in the distribution of observations in the Ramachandran plot of each χ1 rotamer (marked in the figure). At these positions, the Ramachandran populations of the rotamers are significantly reduced. They can be summarized as follows:
| Rotamer | N(i+1) | O(i) |
|---|---|---|
| g+ | ψ = -60° | ψ = +120° |
| trans | ψ = 180° | ψ = 0° |
| Rotamer | C(i-1) | HBond to NH(i) |
| g+ | φ = +60° | φ = -120° |
| g- | φ = -180° | φ = 0° |

Side-chain types with two heavy atoms (Val, Ile, Thr) have backbone-dependent interactions with both heavy atoms. Val has CG1 at χ1 and CG2 at χ1+120°. Because Val g+ and g- conformations have steric interactions with the backbone near ψ=120° and -60° (the most populated ψ ranges), Val is the only amino acid where the t rotamer (χ1~180°) is the most common. At most values of φ and ψ, only one rotamer of Val is allowed (shown in figure). Ile has CG1 at χ1 and CG2 at χ1-120°. Thr has OG1 at χ1 and CG2 at χ1-120°.
Uses
The Dunbrack backbone-dependent rotamer library is used in a number of programs for protein structure prediction and computational design, including:
- Side-chain conformation prediction in protein structure modeling
- Visualization of Protein Mutations
References
- Huang, X; Pearce, R; Zhang, Y (2020). "Toward the Accuracy and Speed of Protein Side-Chain Packing: A Systematic Study on Rotamer Libraries". Journal of Chemical Information and Modeling. 60 (1): 410–420. doi:10.1021/acs.jcim.9b00812. PMC 7938712. PMID 31851497. Retrieved 18 February 2021.
- Dunbrack, RL, Jr.; Karplus, M (1993). "Backbone-dependent rotamer library for proteins. Application to side-chain prediction". Journal of Molecular Biology. 230 (2): 543–74. doi:10.1006/jmbi.1993.1170. PMID 8464064.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Dunbrack, RL, Jr.; Cohen, FE (1997). "Bayesian statistical analysis of protein side-chain rotamer preferences". Protein Science. 6 (8): 1661–81. doi:10.1002/pro.5560060807. PMC 2143774. PMID 9260279.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Bower, MJ; Cohen, FE; Dunbrack, RL, Jr (1997). "Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: a new homology modeling tool". Journal of Molecular Biology. 267 (5): 1268–82. doi:10.1006/jmbi.1997.0926. PMID 9150411.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Kuhlman, B; Baker, D (2000). "Native protein sequences are close to optimal for their structures". Proceedings of the National Academy of Sciences of the United States of America. 97 (19): 10383–8. Bibcode:2000PNAS...9710383K. doi:10.1073/pnas.97.19.10383. PMC 27033. PMID 10984534.
- Dunbrack, RL, Jr (2002). "Rotamer libraries in the 21st century". Current Opinion in Structural Biology. 12 (4): 431–40. doi:10.1016/s0959-440x(02)00344-5. PMID 12163064.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Alford, RF; Leaver-Fay, A; Jeliazkov, JR; O'Meara, MJ; DiMaio, FP; Park, H; Shapovalov, MV; Renfrew, PD; Mulligan, VK; Kappel, K; Labonte, JW; Pacella, MS; Bonneau, R; Bradley, P; Dunbrack, RL; Das, R; Baker, D; Kuhlman, B; Kortemme, T; Gray, JJ (13 June 2017). "The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design". Journal of Chemical Theory and Computation. 13 (6): 3031–3048. doi:10.1021/acs.jctc.7b00125. PMC 5717763. PMID 28430426.
- Shapovalov, MV; Dunbrack, RL, Jr (2011). "A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions". Structure. 19 (6): 844–58. doi:10.1016/j.str.2011.03.019. PMC 3118414. PMID 21645855.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Towse, Clare-Louise; Rysavy, Steven J.; Vulovic, Ivan M.; Daggett, Valerie (5 January 2016). "New Dynamic Rotamer Libraries: Data-Driven Analysis of Side-Chain Conformational Propensities". Structure. 24 (1): 187–199. doi:10.1016/j.str.2015.10.017. ISSN 0969-2126. PMC 4715459. PMID 26745530.
- Levitt, Michael; Hirshberg, Miriam; Sharon, Ruth; Daggett, Valerie (1995). "Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution". Computer Physics Communications. 91 (1): 215–231. Bibcode:1995CoPhC..91..215L. doi:10.1016/0010-4655(95)00049-L.
- Dunbrack, RL, Jr.; Karplus, M (1994). "Conformational analysis of the backbone-dependent rotamer preferences of protein sidechains". Nature Structural Biology. 1 (5): 334–340. doi:10.1038/nsb0594-334. ISSN 1545-9985. PMID 7664040. S2CID 9157373.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Waterhouse, A; Bertoni, M; Bienert, S; Studer, G; Tauriello, G; Gumienny, R; Heer, FT; de Beer, TAP; Rempfer, C; Bordoli, L; Lepore, R; Schwede, T (2018). "SWISS-MODEL: homology modelling of protein structures and complexes". Nucleic Acids Research. 46 (W1): W296–W303. doi:10.1093/nar/gky427. PMC 6030848. PMID 29788355.
- Studer, G; Tauriello, G; Bienert, S; Biasini, M; Johner, N; Schwede, T (2021). "ProMod3-A versatile homology modelling toolbox". PLOS Computational Biology. 17 (1): e1008667. Bibcode:2021PLSCB..17E8667S. doi:10.1371/journal.pcbi.1008667. PMC 7872268. PMID 33507980.
- Kelley, LA; Mezulis, S; Yates, CM; Wass, MN; Sternberg, MJ (2015). "The Phyre2 web portal for protein modeling, prediction and analysis". Nature Protocols. 10 (6): 845–58. doi:10.1038/nprot.2015.053. PMC 5298202. PMID 25950237.
- OpenEye Scientific Software. "Macromolecule Conformations — Toolkits -- Java". OEChem Toolkit 3.1.0.0. Retrieved 10 February 2021.
- Krieger, E. "Protein side-chain modeling in YASARA". www.yasara.org. Yasara Biosciences. Retrieved 10 February 2021.
- Heo, L; Park, H; Seok, C (2013). "GalaxyRefine: Protein structure refinement driven by side-chain repacking". Nucleic Acids Research. 41 (Web Server issue): W384-8. doi:10.1093/nar/gkt458. PMC 3692086. PMID 23737448.
- Krivov, Georgii G.; Shapovalov, Maxim V.; Dunbrack, Roland L. (2009). "Improved prediction of protein side-chain conformations with SCWRL4". Proteins: Structure, Function, and Bioinformatics. 77 (4): 778–795. doi:10.1002/prot.22488. ISSN 1097-0134. PMC 2885146. PMID 19603484.
- Huang, X; Pearce, R; Zhang, Y (2020). "EvoEF2: accurate and fast energy function for computational protein design". Bioinformatics. 36 (4): 1135–1142. doi:10.1093/bioinformatics/btz740. PMC 7144094. PMID 31588495.
- Kulp, Daniel W. "Rotamer Toggle - PyMOLWiki". pymolwiki.org. SBGrid Consortium. Retrieved 10 February 2021.
- Pettersen, EF; Goddard, TD; Huang, CC; Meng, EC; Couch, GS; Croll, TI; Morris, JH; Ferrin, TE. "Rotamer Tools (ChimeraX)". The ChimeraX User Guide. Regents of the University of California. Retrieved 9 February 2021.